Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Gabriela Trevisan and Version 2 by Conner Chen.
The superfamily of ion channels named transient receptor potential (TRP) acts as sensors of oxidative compounds at the plasma membrane and can amplify several signaling. The TRP superfamily is a non-selective cation channel initially identified in the Drosophila fly species. These channels are presented in different cell types and tissues, such as epithelial, immune, and neuronal cells.
- chronic pain
- oxidative stress
- TRP channels
- allodynia
1. Neuropathic Pain
Pain can be categorized as acute or chronic pain, where chronic pain is defined by the International Association for the Study of Pain (IASP) as “pain that persists or recurs for more than 3 months”. This pain occurs in approximately 20% of the general population and negatively affects the quality of life, a common cause of disability and suffering in patients [1]. The last revision of the International Classification of Diseases (ICD-11) included a new system for the classification of chronic pain as primary and secondary. The first is detected in patients with migraine, fibromyalgia, or complex regional pain syndrome. This type of chronic pain cannot be explained by a complication of another chronic pain condition. In contrast, secondary chronic pain arises initially as a symptom of another disease, such as cancer pain and diabetic polyneuropathy [2].
Neuropathic pain is a frequent type of chronic pain, affecting 7–10% of people worldwide. However, the incidence of neuropathic pain probably will enhance with the growing number of diagnoses for diabetes mellitus, the aging population, and chemotherapy for cancer treatment. The IASP defines this form of pain as “pain caused by a lesion or disease of the somatosensory nervous system” [1]. Additionally, it is classified as central pain (in multiple sclerosis, central post-stroke pain, and spinal cord injury) or peripheral pain (detected in diabetic polyneuropathy, chemotherapeutic administration, and peripheral nerve trauma). Patients often describe neuropathic pain as paresthesia, spontaneous (pricking or burning), or evoked pain (such as allodynia and hyperalgesia to mechanical or thermal painful stimuli). Moreover, negative sensory symptoms may also be present in neuropathic pain, including sensory loss and numbness [3][4].
The current pharmacological treatment for neuropathic pain involves medicines that only control painful symptoms. Then, distinct analgesics can be used as first-line therapies, including gabapentinoids (pregabalin and gabapentin), tricyclic antidepressants, and duloxetine (selective serotonin and norepinephrine reuptake inhibitor antidepressant, SSNRI). Alternative solutions should be used to manage neuropathic pain, such as high-concentration capsaicin patches (a transient receptor potential vanilloid 1–TRPV1 agonist found in chili peppers) and tramadol (an opioid agonist that also can act as a SSNRI) [5], although these compounds have adverse effects and still show limited efficacy in controlling neuropathic pain [3][4].
In this view, studying the mechanisms involved in neuropathic pain development could help design better therapeutic options and, in some cases of neuropathic pain, propose a way to prevent it, such as the neuropathic pain caused by chemotherapeutic administration or diabetes. Different mechanisms may lead to neuropathic pain induction, such as altered ion channel expression and function when there is a nerve injury or a disease of the somatosensory system. In addition, oxidative compounds can also be involved in neuropathic pain development [6]. Then, this process may cause increased excitation of the peripheral and central nervous nociceptive neurons or a reduction in the inhibition of ascending afferent pathways. The superfamily of ion channels named transient receptor potential (TRP) acts as sensors of oxidative compounds at the plasma membrane and can amplify several signaling pathways [7]. Therefore, specific TRP modulators, such as agonists, antagonists, and endogenous modulators, have been identified and analyzed for future therapeutic applications to treat chronic diseases, such as neuropathic pain [3][4][8].
2. Transient Receptor Potential (TRP)
The TRP superfamily is a non-selective cation channel initially identified in the Drosophila fly species. These channels are presented in different cell types and tissues, such as epithelial, immune, and neuronal cells. In mammals, the TRP superfamily has 28 described members, which are divided into six subgroups according to channel structure: TRP Canonical (TRPC), TRP Vanilloid (TRPV), TRP Melastatin (TRPM), TRP Polycystin (TRPP), TRP Mucolipin (TRPML) and TRP Ankyrin (TRPA) [7]. TRP channels are activated by pharmacological ligands, temperature, pH, and mechanical force [9]. Additionally, inflammatory and oxidative compounds generated after tissue lesions can activate or sensitize these channels [10]. After activation, the TRP channels mediate the influx of different ions, such as sodium (Na+), calcium (Ca2+) and magnesium (Mg2+) [7].
TRP channels share a typical structure of six transmembrane regions with amino and carboxyl terminus in the cytoplasm [9]. The first member of the TRP channel family that has its structure completely elucidated was TRPV1, followed by TRPA1 and transient potential receptor vanilloid 4 (TRPV4) [11]. The structural elucidation helped the investigation of their involvement in pathologies, such as pain, chronic inflammation, fibrosis and edema [12]. Moreover, TRPA1, TRPV1 and TRPV4 channels co-localize in primary sensory neurons and are related to inflammatory and neuropathic pain development in pre-clinical models [8].
Among the six family members, TRPV1–4 are thermosensitive, while TRPV5–6 are not sensitive to temperature [13]. TRPV1 is activated by capsaicin and noxious temperatures (≥43 °C) and is associated with neuropathic and inflammatory pain induction [13]. Capsaicin is the only drug approved for clinical use that binds to TRP channels (TRPV1) and can be used, as a second-line drug, for diabetic peripheral neuropathy treatment (Qutenza®). Additionally, clinical trials have demonstrated capsaicin efficacy for post-herpetic neuralgia, post-traumatic or post-surgical nerve injury, human immunodeficiency virus (HIV)-induced neuropathy and chronic painful chemotherapy. However, capsaicin can only be used for topical application (local pharmacological action) due to the side effects, such as erythema, pruritus, reduced heat sensitivity and pain [14]. Therefore, TRP channels still need to be investigated as new pharmacological targets to treat neuropathic pain.
In this view, TRPV4 has been widely studied and linked to many channelopathies, suggesting a broad expression pattern and versatile physiological function [15]. The TRPV4 gene mutations have generated osteoarthropathy, skeletal dysplasia and peripheral neuropathies, manifesting as a variable combination of skeletal, motor and neuronal symptoms, including pain [16]. Therefore, the TRPV4 channel is particularly interesting due to its involvement in neuropathic pain symptoms [8].
 Furthermore, when TRPV4 is activated, Ca2+ influx enhances the signaling of protein kinase C (PKC)-dependent phosphorylation. The PKC activation in the dorsal root ganglion (DRG) neurons generates TRPV4 sensitization and plays a role in nociception [23]. Another signaling pathway that activates TRPV4 is protease activating receptor 2 (PAR2), probably by PKC and protein kinase A (PKA) activation. The PAR2 is a G protein-coupled receptor that is expressed in alveolar macrophages, endothelial cells, and epithelial cells. This receptor can modulate inflammatory responses due to pro-inflammatory cytokines production, such as interleukin-1β (IL-1β), interleukin-6 (IL-6) and interleukin-8 (IL-8), which are involved in pain control [18].
In addition, nitric oxide (NO), has been associated with mechanisms of neuropathic pain and peripheral nerve injury, possibly due to the modification of protein kinase and ion channels [24]. The TRPV4-mediated Ca2+ influx can activate the inducible nitric oxide synthase (iNOS)-nitric oxide, which increases nitric oxide (NO)NO release. NO can activate the cyclic adenosine monophosphate (cAMP)-dependent PKA and cyclic guanosine monophosphate (cGMP)-dependent protein kinase G (PKG), which contribute to hyperalgesia (NO-cGMP-PKG) [25]. This activation induces the signaling mechanism through second messengers such as mitogen-activated protein kinases (MAPK) and the nuclear factors kappa B (NF-κB) [25]. This pathway activation was reported in diabetic neuropathy, paclitaxel-induced peripheral neuropathy, and nerve injury [26][27][28]. Therefore, a pharmacological approach that induces TRPV4 downregulation could decrease neuropathic pain due to a reduction in NO production [25]. Then, TRPV4 activation may be mediated by direct agonist production or be sensitized by diverse mechanisms.
Recently, it has been described that the GSK2798745 (TRPV4 antagonist), a new TRPV4 antagonist, has been well-tolerated in healthy volunteers in Phase I clinical trials [22]. Therefore, this compound administration did not induce adverse effects, suggesting that TRPV4 antagonists could be a potential therapeutic target in pain conditions, such as inflammatory pain, neuropathic pain, cancer pain, and migraines. The TRPV4 expressions in the DRG, peripheral fibers, and spinal cord of healthy patients were observed by immunohistochemistry. However, in diabetic patients with neuropathy, the TRPV4 expression did not change in the skin nerve fibers. The non-increased TRPV4 expression suggests the agonist effect on constitutive receptors possibly generated neuropathic pain.
Furthermore, the TRPV4 knockout (Trpv4−/−) mice did not show impaired heat or touch sensation. However, in inflammatory pain, mechanical allodynia, hyposmotic solution-induced nociception, edema formation and cytokine release were reduced in these animals [29]. In addition, a fly model of neuropathy observed that mutations within the TRPV4 cause disruption of axonal interactions and dendritic degeneration [30]. The TRPV4 role has been researched in models that induce neuropathic pain, such as trauma, surgery, chemotherapy, diabetes and alcohol intake [31][32][33][34][35].
Furthermore, when TRPV4 is activated, Ca2+ influx enhances the signaling of protein kinase C (PKC)-dependent phosphorylation. The PKC activation in the dorsal root ganglion (DRG) neurons generates TRPV4 sensitization and plays a role in nociception [23]. Another signaling pathway that activates TRPV4 is protease activating receptor 2 (PAR2), probably by PKC and protein kinase A (PKA) activation. The PAR2 is a G protein-coupled receptor that is expressed in alveolar macrophages, endothelial cells, and epithelial cells. This receptor can modulate inflammatory responses due to pro-inflammatory cytokines production, such as interleukin-1β (IL-1β), interleukin-6 (IL-6) and interleukin-8 (IL-8), which are involved in pain control [18].
In addition, nitric oxide (NO), has been associated with mechanisms of neuropathic pain and peripheral nerve injury, possibly due to the modification of protein kinase and ion channels [24]. The TRPV4-mediated Ca2+ influx can activate the inducible nitric oxide synthase (iNOS)-nitric oxide, which increases nitric oxide (NO)NO release. NO can activate the cyclic adenosine monophosphate (cAMP)-dependent PKA and cyclic guanosine monophosphate (cGMP)-dependent protein kinase G (PKG), which contribute to hyperalgesia (NO-cGMP-PKG) [25]. This activation induces the signaling mechanism through second messengers such as mitogen-activated protein kinases (MAPK) and the nuclear factors kappa B (NF-κB) [25]. This pathway activation was reported in diabetic neuropathy, paclitaxel-induced peripheral neuropathy, and nerve injury [26][27][28]. Therefore, a pharmacological approach that induces TRPV4 downregulation could decrease neuropathic pain due to a reduction in NO production [25]. Then, TRPV4 activation may be mediated by direct agonist production or be sensitized by diverse mechanisms.
Recently, it has been described that the GSK2798745 (TRPV4 antagonist), a new TRPV4 antagonist, has been well-tolerated in healthy volunteers in Phase I clinical trials [22]. Therefore, this compound administration did not induce adverse effects, suggesting that TRPV4 antagonists could be a potential therapeutic target in pain conditions, such as inflammatory pain, neuropathic pain, cancer pain, and migraines. The TRPV4 expressions in the DRG, peripheral fibers, and spinal cord of healthy patients were observed by immunohistochemistry. However, in diabetic patients with neuropathy, the TRPV4 expression did not change in the skin nerve fibers. The non-increased TRPV4 expression suggests the agonist effect on constitutive receptors possibly generated neuropathic pain.
Furthermore, the TRPV4 knockout (Trpv4−/−) mice did not show impaired heat or touch sensation. However, in inflammatory pain, mechanical allodynia, hyposmotic solution-induced nociception, edema formation and cytokine release were reduced in these animals [29]. In addition, a fly model of neuropathy observed that mutations within the TRPV4 cause disruption of axonal interactions and dendritic degeneration [30]. The TRPV4 role has been researched in models that induce neuropathic pain, such as trauma, surgery, chemotherapy, diabetes and alcohol intake [31][32][33][34][35].
Transient Receptor Potential Vanilloid 4 (TRPV4)
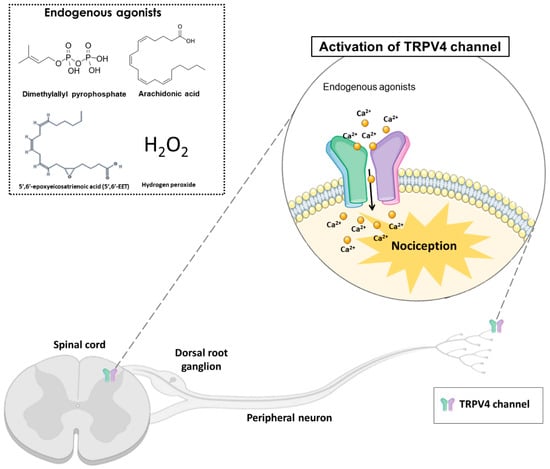
The TRPV4 channel structure contains six transmembrane regions (S1–S6), similar to other TRP proteins. The intracellular N-region is linked to the S1 region and includes six ankyrin domains, a proline-rich domain and a linker domain with two β-strands [17]. The TRP helix pore-forming loop allows the ionic flow and is located between S5 and S6 domains. Additionally, extending from the S6 transmembrane region is observed the C-terminal domain with a folding recognition domain (FRD), a TRP domain and a PDZ-binding domain [17]. These structure characteristics are essential for assembly and localization, possibly contributing to the activation and modulation of the TRPV4 function [18]. The X-ray crystallographic analyses identify that the symmetric tetramer architecture of TRPV4 is similar to other TRPV channels, including TRPV1, TRPV2 and TRPV6. However, the unique arrangement of the S1–S4 domain packing against the S5–S6 transmembrane domains differentiated this channel from the other TRPVs. Thus, clarifying the molecular and cellular mechanisms underlying the TRPV4 channel is fundamental to understanding channel function and designing effective therapies [19]. The TRPV4 channel is expressed in immune cells, such as macrophages, neutrophils, and dendritic cells. Moreover, this channel is also expressed in sensory neurons, glial cells, the spinal cord, cortical pyramidal neurons, the thalamus, and cerebellum basal nuclei [20]. Thus, this channel is associated with inflammatory diseases that affect the central and peripheral nervous system, such as osteoarthritis, atherosclerosis, cancer pain, and neuropathies [21]. TRPV4 can be activated by hyposmolarity, non-noxious heat (38 °C), hydrogen peroxide (H2O2), low pH (5.0), mechanical forces, and ultraviolet B-rays (UVB) radiation. Additionally, exogenous or endogenous chemical compounds can activate or block the TRPV4 channel. The TRPV4 endogenous agonists include arachidonic acid, 5′,6′-epoxyeicosatrienoic acid (5′,6′-EET) and dimethylallyl pyrophosphate (Figure 1). In addition, the exogenous agonists include x-3 polyunsaturated fatty acids, bisandrographolide A, 4α-phorbol 12,13-didecanoate (4α-PDD) and GSK1016790A [17]. Although the therapeutic potential of selective TRPV4 agonists has been hypothesized, most clinical interest has concentrated on channel inhibition [22]. In this context, TRPV4 antagonists such as HC-067047, RN-1734 and GSK2193874 have been evaluated and used in pre-clinical models to reduce nociception [22].Figure 1. The activation of transient receptor potential vanilloid 4 (TRPV4) by endogenous agonists (dimethylallyl pyrophosphate, arachidonic acid, 5′,6′-epoxyeicosatrienoic acid, and hydrogen peroxide) in the dorsal spinal cord and peripheral nociceptive neurons, may cause nociception in different pain models. TRPV4 is a non-selective calcium channel that mediates calcium influx, promoting nociception transduction.
References
- Treede, R.-D.; Rief, W.; Barke, A.; Aziz, Q.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Evers, S.; Finnerup, N.B.; First, M.B.; et al. Chronic pain as a symptom or a disease: The IASP Classification of Chronic Pain for the International Classification of Diseases (ICD-11). Pain 2019, 160, 19–27.
- Scholz, J.; Finnerup, N.B.; Attal, N.; Aziz, Q.; Baron, R.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Cruccu, G.; Davis, K.D.; et al. The IASP classification of chronic pain for ICD-11: Chronic neuropathic pain. Pain 2019, 160, 53–59.
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Prim. 2017, 3, 17002.
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic Pain: From Mechanisms to Treatment. Physiol. Rev. 2021, 101, 259–301.
- Yamasaki, H.; Funai, Y.; Funao, T.; Mori, T.; Nishikawa, K. Effects of Tramadol on Substantia Gelatinosa Neurons in the Rat Spinal Cord: An In Vivo Patch-Clamp Analysis. PLoS ONE 2015, 10, e0125147.
- Dalenogare, D.P.; Theisen, M.C.; Peres, D.S.; Fialho, M.F.P.; Luckemeyer, D.D.; Antoniazzi, C.T.D.; Kudsi, S.Q.; Ferreira, M.A.; Ritter, C.D.S.; Ferreira, J.; et al. TRPA1 activation mediates nociception behaviors in a mouse model of relapsing-remitting experimental autoimmune encephalomyelitis. Exp. Neurol. 2020, 328, 113241.
- Koivisto, A.P.; Belvisi, M.G.; Gaudet, R.; Szallasi, A. Advances in TRP channel drug discovery: From target validation to clinical studies. Nat. Rev. Drug Discov. 2022, 21, 41–59.
- Iannone, L.F.; De Logu, F.; Geppetti, P.; De Cesaris, F. The role of TRP ion channels in migraine and headache. Neurosci. Lett. 2022, 768, 136380.
- Gu, Q.; Lee, L.Y. TRP channels in airway sensory nerves. Neurosci. Lett. 2021, 748, 135719.
- Talavera, K.; Startek, J.B.; Alvarez-Collazo, J.; Boonen, B.; Alpizar, Y.A.; Sanchez, A.; Naert, R.; Nilius, B. Mammalian Transient Receptor Potential TRPA1 Channels: From Structure to Disease. Physiol. Rev. 2020, 100, 725–803.
- Moran, M.M. TRP Channels as Potential Drug Targets. Annu. Rev. Pharm. Toxicol. 2018, 58, 309–330.
- Dietrich, A. Transient Receptor Potential (TRP) Channels in Health and Disease. Cells 2019, 8, 413.
- Pumroy, R.A.; Fluck, E.C., 3rd; Ahmed, T.; Moiseenkova-Bell, V.Y. Structural insights into the gating mechanisms of TRPV channels. Cell Calcium 2020, 87, 102168.
- Arora, V.; Campbell, J.N.; Chung, M.K. Fight fire with fire: Neurobiology of capsaicin-induced analgesia for chronic pain. Pharmacol. Ther. 2021, 220, 107743.
- Seebohm, G.; Schreiber, J.A. Beyond Hot and Spicy: TRPV Channels and their Pharmacological Modulation. Cell Physiol. Biochem. 2021, 55, 108–130.
- Taga, A.; Peyton, M.A.; Goretzki, B.; Gallagher, T.Q.; Ritter, A.; Harper, A.; Crawford, T.O.; Hellmich, U.A.; Sumner, C.J.; McCray, B.A. TRPV4 mutations causing mixed neuropathy and skeletal phenotypes result in severe gain of function. Ann. Clin. Transl. Neurol. 2022, 9, 375–391.
- Ji, C.; McCulloch, C.A. TRPV4 integrates matrix mechanosensing with Ca(2+) signaling to regulate extracellular matrix remodeling. FEBS J. 2021, 288, 5867–5887.
- Yu, S.; Huang, S.; Ding, Y.; Wang, W.; Wang, A.; Lu, Y. Transient receptor potential ion-channel subfamily V member 4: A potential target for cancer treatment. Cell Death Dis. 2019, 10, 497.
- Deng, Z.; Paknejad, N.; Maksaev, G.; Sala-Rabanal, M.; Nichols, C.G.; Hite, R.K.; Yuan, P. Cryo-EM and X-ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms. Nat. Struct. Mol. Biol. 2018, 25, 252–260.
- Kumar, H.; Lee, S.-H.; Kim, K.-T.; Zeng, X.; Han, I. TRPV4: A Sensor for Homeostasis and Pathological Events in the CNS. Mol. Neurobiol. 2018, 55, 8695–8708.
- Nguyen, T.N.; Siddiqui, G.; Veldhuis, N.A.; Poole, D.P. Diverse Roles of TRPV4 in Macrophages: A Need for Unbiased Profiling. Front. Immunol. 2021, 12, 828115.
- Lawhorn, B.G.; Brnardic, E.J.; Behm, D.J. TRPV4 antagonists: A patent review (2015–2020). Expert Opin. Ther. Patents 2021, 31, 773–784.
- Costa, R.; Bicca, M.A.; Manjavachi, M.N.; Segat, G.C.; Dias, F.C.; Fernandes, E.S.; Calixto, J.B. Kinin Receptors Sensitize TRPV4 Channel and Induce Mechanical Hyperalgesia: Relevance to Paclitaxel-Induced Peripheral Neuropathy in Mice. Mol. Neurobiol. 2018, 55, 2150–2161.
- Gamper, N.; Ooi, L. Redox and nitric oxide-mediated regulation of sensory neuron ion channel function. Antioxid. Redox Signal. 2015, 22, 486–504.
- Sandireddy, R.; Yerra, V.G.; Areti, A.; Komirishetty, P.; Kumar, A. Neuroinflammation and Oxidative Stress in Diabetic Neuropathy: Futuristic Strategies Based on These Targets. Int. J. Endocrinol. 2014, 2014, 674987.
- Li, J.; Hu, X.; Liang, F.; Liu, J.; Zhou, H.; Liu, J.; Wang, H.; Tang, H. Therapeutic effects of moxibustion simultaneously targeting Nrf2 and NF-κB in diabetic peripheral neuropathy. Appl. Biochem. Biotechnol. 2019, 189, 1167–1182.
- Suo, J.; Wang, M.; Zhang, P.; Lu, Y.; Xu, R.; Zhang, L.; Qiu, S.; Zhang, Q.; Qian, Y.; Meng, J.; et al. Siwei Jianbu decoction improves painful paclitaxel-induced peripheral neuropathy in mouse model by modulating the NF-κB and MAPK signaling pathways. Regen. Med. Res. 2020, 8, 2.
- Lim, H.; Lee, H.; Noh, K.; Lee, S.J. IKK/NF-kappaB-dependent satellite glia activation induces spinal cord microglia activation and neuropathic pain after nerve injury. Pain 2017, 158, 1666–1677.
- Grace, M.S.; Bonvini, S.J.; Belvisi, M.G.; McIntyre, P. Modulation of the TRPV4 ion channel as a therapeutic target for disease. Pharmacol. Ther. 2017, 177, 9–22.
- McCray, B.A.; Diehl, E.; Sullivan, J.M.; Aisenberg, W.H.; Zaccor, N.W.; Lau, A.R.; Rich, D.J.; Goretzki, B.; Hellmich, U.A.; Lloyd, T.E.; et al. Neuropathy-causing TRPV4 mutations disrupt TRPV4-RhoA interactions and impair neurite extension. Nat. Commun. 2021, 12, 1444.
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado, J.D.D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-De-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342.
- Maqboul, A.; Elsadek, B. Expression profiles of TRPV1, TRPV4, TLR4 and ERK1/2 in the dorsal root ganglionic neurons of a cancer-induced neuropathy rat model. PeerJ 2018, 6, e4622.
- Dias, F.C.; Alves, V.S.; Matias, D.O.; Figueiredo, C.P.; Miranda, A.L.P.; Passos, G.F.; Costa, R. The selective TRPV4 channel antagonist HC-067047 attenuates mechanical allodynia in diabetic mice. Eur. J. Pharm. 2019, 856, 172408.
- Sanchez, J.C.; Munoz, L.V.; Ehrlich, B.E. Modulating TRPV4 channels with paclitaxel and lithium. Cell Calcium 2020, 91, 102266.
- Liu, W.; Pu, B.; Liu, M.; Zhang, X.; Zeng, R. Down-regulation of MAPK pathway alleviates TRPV4-mediated trigeminal neuralgia by inhibiting the activation of histone acetylation. Exp. Brain Res. 2021, 239, 3397–3404.
More