Variation in chromosome structure is a central source of DNA damage and DNA damage response, together representinga major hallmark of chromosomal instability. Treatment of cancer usually includes surgery, radiotherapy, or chemotherapy. The activity of radiotherapy and most chemotherapy drugs induces DNA damage in proliferating tumor cells. For example, platinum drugs that are effective against cancer cells form DNA adducts. Other drugs, such as anthracyclines induce DNA damage by inhibiting DNA topoisomerases and promoting the production of mitochondrial reactive oxygen species (ROS). In normal cells, DNA damage can be immediately recognized by a DNA damage response (DDR), which activates checkpoints for cell-cycle arrest and DNA repair. In cancer cells, DNA damage per se may be responsible for defective chromosome segregation and chromosomal instability (CIN) or the anomalous binding of DDR proteins to DNA or chromatin proteins.
1. DNA Damage and Chromosomal Instability
Exogenous and endogenous insults can cause DNA damage. Ionizing radiation (IR) and chemotherapeutic agents, particularly lethal to cancer cells, cause DNA double-strand breaks (DSBs); cell metabolism products (e.g., reactive oxygen species) can cause DSBs as well, but it is also clear that stress replication and chromosome mis-segregation provoke DNA damage.Moreover, chromosomes containing damaged DNA segregate inappropriately because sister chromatids are still connected by DNA and/or DNA-proteins during the division of DNA into daughter cells
[1][2][3]. In anaphase, defective chromosome segregation can produce lagging chromosomes or bridge chromosomes.Indeed, errors in the function of the mitotic spindle can produce lagging chromosomes such that the entire chromosome cannot be properly separated due to the connection with the microtubules derived from the opposite spindle poles
[4][5][6]. Anaphase spindles working inappropriately may also induce bridged chromatin in such a way that DNA is extended to opposite spindle poles from the same chromosome or non-disjoined sister chromatids.Chromatin bridges are hallmarks of structural chromosomal instability.Noteworthily, lagging chromosomes and chromatin bridges often do not catch up with the other chromosomes to be introduced in the reforming nucleus. This chromatin self-organizes into micronuclei surrounded by a nuclear membrane (
Figure 1). The level of DNA damage in micronuclei is high, and the repair of this damage leads to extensive gene rearrangements
[7].
Figure 1. Chromosome mis-segregation causes DNA damage. (A) Precise chromosome segregation leads to the equal partitioning of the genome and the generation of two euploid daughter cells with diploid karyotype (illustrated as 2N). (B) Defective chromosome segregation can have multiple fates. Chromosomes can be trapped in the cytokinetic furrow and break during cytokinesis (top). Alternatively, chromosomes can mis-segregate and form micronuclei (middle), or they can be accurately segregated in nucleus/micronucleus in the daughter cells (bottom). Irrespective of how micronuclei are generated, their DNA is significantly damaged and elicits a DNA damage response.
2. Importance of DNA DRamage Response and Chromatin Remodeling in Therapeutics Response and Resistance
In response to therapeutic intervention, cancer cells can redirect DNA repair pathways. NHEJ, which is the prevalent DSB repair pathway in higher eukaryotes, essentially mediates the direct ligation of broken DNA ends and usually involves minimal DNA end processing. Changes in the balance between HR and NHEJ, for example, can alter the DNA repair-deficient tumor cell response to DSB-inducing drugs (
Figure 2). Although there is no error in HR, NHEJ can cause mutations that are favorable for survival. Hence, many tumor resistances lacking HR can be explained by abnormal regulation of NHEJ activity. Tumors with HR defects, through the rewiring of DNA repair pathways, will thwart the therapeutic response. For instance, as aforementioned, loss of 53BP1 rescues cancer cells with BRCA1 deficiency, including HR impairment and chemotherapeutic sensitivity
[8][9][10]. Similarly, overexpression of mutant 53BP1 inhibits NHEJ and stimulates HR in an XRCC4-dependent manner. A similar change from NHEJ to HR was also found in embryonic stem cells deficient in XRCC4 or other NHEJ proteins
[11][12]. The blocking of NHEJ by Lig4 deletion did not, however, rescue BRCA1 mutant cells from genomic instability. The results indicate that the HR recovery caused by the loss of 53BP1 is an important factor in the chemotherapy response
[9][10]. Therefore, since the abnormal expression of 53BP1 recurs in a variety of tumors and is associated with decreased survival without metastasis, these aspects may be clinically important. As has been shown for BRCA1- and BRCA2- mutated tumors, drugs targeting D
NA damage response (D
RDR) may select for secondary mutations, which result in partial or complete restoration of protein function. These secondary mutations may lead to alternative splicing, losing the mutant exon, and causing a second frameshift mutation that restores the reading frame
[13][14][15].
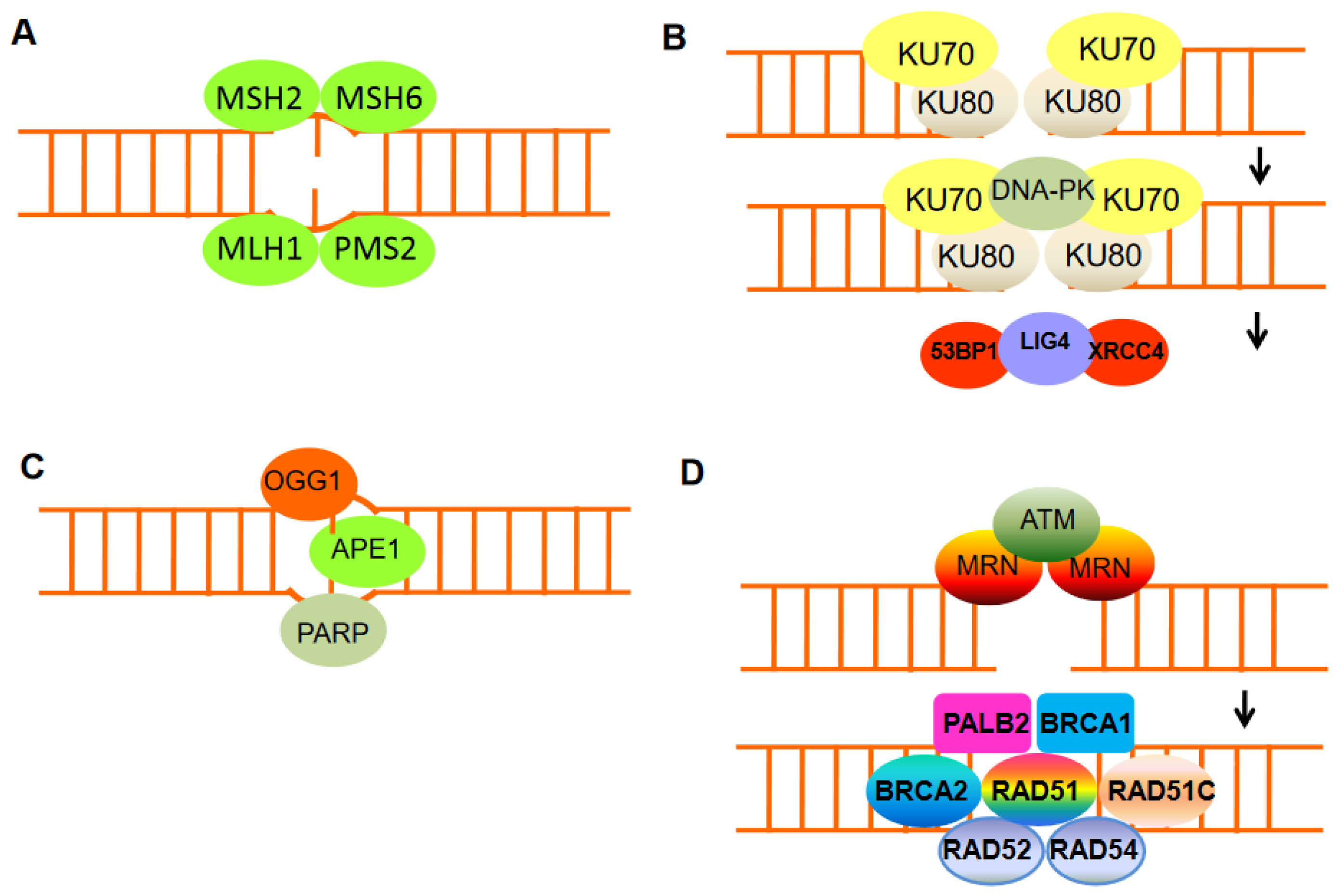
Figure 2. The mechanisms of DNA damage repair. (A) Mismatch repair proteins localize and repair mismatched bases. (B) Nonhomologous end-joining (NHEJ) requires a nuclease to resect damaged DNA, DNA polymerases to fill in new DNA, and a ligase to restore integrity to the DNA strands. The Ku bound to the end of the DNA can be considered as a Ku:DNA complex, which is a node where the nuclease, polymerase, and ligase of NHEJ can dock. DNA-PK has a molecular weight of 469 kDa and contains 4128 aa. It is the largest protein kinase in biology and is the only protein kinase that is specifically activated by binding to the ends of double-stranded DNA with multiple end configurations. Activated DNA-PK stimulates the ligase activity of XRCC4: DNA ligase 4. (C) Base excision repair can remove damaged DNA bases, e.g., by oxidation, using damage-specific DNA glycosylase, in the case of oxidizing guanine, 8-oxoguanine DNA glycosylase 1 (OGG1). (D) DNA double-strand break repair by homologous recombination (HR) can only be active in late S or G2 phases of the cell cycle when sister chromatid templates are available. HR includes DNA excision through MRN, homologous pairing and strand exchange through RAD51 and RAD52, Rad51c, Rad54, and BRCA1–PALB2, and DNA synthesis.
The newly discovered field of translesion synthesis (TLS) has suggested that mammalian cells need distinct polymerases to efficiently and accurately bypass DNA lesions. Translesion synthesis is the newest and less characterized pathway of DNA repair. It involves DNA polymerases that facilitate DNA replicationby efficiently bypassing various DNA lesions in a relatively error-free manner. TLS polymerase gene expression, mutation, and regulation are important for cancer etiology and treatment.These polymerases are enzymes with low fidelity when copying undamaged DNA; however, they copy damaged DNA with much higher efficiency compared to the replicative DNA enzymes
[16]. These findings have led to a model proposing that, during DNA replication, the proofreading DNA polymerase gets stalled at sites of DNA damage, and it is replaced by a low-fidelity DNApolymerase. The perturbation due to not proofreading DNA polymerase by mutation or loss of expressionis expected to result in the accumulation of mutations in cells exposed to specific carcinogens; therefore, TLS genes and TLS gene variations represent attractive pharmacologic targets
[16].
In the case of the DNA mismatch repair (MMR) system, other resistance mechanisms are involved. To maintain genome integrity, the MMR system is essential, and mutations in MMR genes (such as MLH1 and MSH2) can lead to a microsatellite instability (MSI) phenotype
[17]. MMR deficiency is associated with tolerance to different cytotoxic chemotherapies (e.g., doxorubicin); furthermore, resistance to cisplatin and carboplatin has been reported to cause hypermethylation of MLH1. However, a subset of cancers characterized by high tumor DNA damage and high susceptibility to immune checkpoint inhibitors can be identified by the increased level of microsatellite instability. Interestingly, cancers associated with defective MMR do not display the mutational disparity between regions with high and low histone H3 lysine 9 methyl 3 (H3K9me3). This loss of mutational disparity in MMR-deficient cancers is due to increased mutation rates in regions lacking H3K9me3. Thus, the higher recovery of mutations in H3K9me3 domains observed in MMR-competent cancer cells is likely caused by reduced MMR repair rates in high H3K9me3 regions. A change in histone modifications can directly affect DNA damage repair efficiency since many histone modifications have been implicated in promoting or inhibiting the recruitment of specific repair proteins
[18][19].
Maintenance of genomic stability in cells requires a tight regulation of histone modifications that accompany DDR. A long-standing question in the study of the cellular response to DNA damage is how the complex structural environment of chromatin is altered in the presence of DNA lesions
[18]. Although DNA damage repair mechanisms have been extensively studied, the exact order of histone modifier and remodeler recruitment remains obscure. It seems more evident that a number of recruited factors regulate each other’s accumulation and activation, rendering the study of the specific function of each factor more difficult. The basic repeating unit of chromatin, the nucleosome, consists of DNA wrapped around an octamer of core histones, which is composed of two molecules each of the histones H2A, H2B, H3, and H4. Nucleosomal DNA is dynamically packaged to varying degrees, resulting in different levels of chromatin compaction ranging from the 10nm fiber to higher-order structures such as the condensed mitotic chromosomes. Heterochromatin plays a major role in the maintenance of the genome stability by repressing transposable elements including DNA transposons and retrotransposons (RTEs), further classified as LTR sequences that characterize endogenous retroviruses (ERVs) and non-LTR elements such as long or short interspersed elements. Propagation of RTEs in the genome has been recognized as a great source of genomic instability
[19]. Heterochromatin is classically described as a condensed, densely stained region of DNA that contains few active genes but is enriched for repetitive sequences. Mammalian heterochromatin is characterized by high levels of the histone modifications H3K9me3 and H3K27me3 and low levels of histone acetylation. Heterochromatin is maintained by a dense array of specific chromatin-binding proteins, including members of the HP1 family (which bind to methylated H3K9), histone deacetylases (HDACs), and histone methyltransferases. The importance of chromatin organization in maintaining genomic stability is underscored by studies of sequencing of multiple cancer genomes; these studies have revealed that mutations accumulate at much higher levels in compact, H3K9me3-rich heterochromatin domains
[20][21]. Furthermore, insertion and deletions are localized around nucleosomes, whereas mutations tend to cluster on the nucleosomal DNA
[22], and both can be influenced by the presence of specific epigenetic modifications on the nucleosome
[20]. Some of these differences in mutation rates may occur via negative selection or through protection of DNA from mutagens by association with nucleosomes. However, the elevated mutation rates in compact, transcriptionally silent heterochromatin domains imply that chromatin packing may impact the detection or repair of damage by the DDR machinery. That is, the ability of the DDR machinery to access the DNA can have a significant impact on genomic stability within specific regions. Indeed, heterochromatin dysfunction provokes genetic instability by inducing aberrant repeat repair, chromosome segregations errors, and replication stress. The therapeutic potential of combination therapy to cope resistance relies on the simultaneous targeting of several different hallmarks of cancer; chromatin remodeling and epigenetic factors may influence cancer response to treatment in ways that are complementary to the more common targets of apoptosis and cell proliferation.