1. Introduction
Advanced age is one of the independent risk factors, correlated with multiple disorders such as diabetes, hypertension, heart failure, obesity, and obstructive sleep apnea syndrome (OSAS) [1][3]. Because of the aging of the global population, it is expected to be more prevalent in individuals aged over 65 by 2060 [2][3][4,5]. The interaction of distinct factors, such as inflammation, structural remodeling, fibrosis, and ion-channel dysfunction, guarantees the onset of a complex pathophysiological mechanism that starts atrial fibrillation and worsens structural and electrical changes in the atria [4][2]. The generation of quick multiple ectopic electrical pulses is able to start and sustain irregular electrical activity of atrial fibrillation. The most common locations of occurrence of ectopic foci are the pulmonary veins, whose isolation is the cornerstone of catheter ablation procedures [5][6][6,7], and less commonly the interatrial septum, coronary sinus, and superior vena cava [7][8][8,9]. These electrical alterations also help the AF-associated hypercoagulable state. Failed electrical regularity and excessive ectopic contractility antagonize local atrial hypo contractility, increasing endothelial expression of plasminogen activator inhibitor (PAI-1) [9][10], contributing to clot generation. Various diseases are risk factors for the occurrence of AF, such as hypertension [10][11], heart failure [11][12] and diabetes [12][13][13,14]. Their common factor is represented by the inflammatory state involved in each of these disorders, which plays a key role in the pathophysiology of AF by mediating the production of cytokines and reactive oxygen species that can increase the disease state, fibrosis, and atrial remodeling [14][15]. Hypo contractility, as well as blood stasis, promotes the development of endothelial microdamage, which attracts the migration and infiltration of several cells of the innate immune system, including macrophages and leukocytes. This mechanism damages the atrial architecture, promoting an inflammatory process, remodels the walls of the atrium, fostering fibrous tissue growth and destroying cardiomyocytes, thus increasing the local inflammatory response and helping the expression of endothelial adhesion molecules and inflammatory cytokines [15][16][16,17]. Alarmins represent a group of endogenous molecules characterized by multiple functions. They can be classified into three categories: (1) granule-derived, as α- and β-defensins, cathelicidin (LL37/cathelicidin-related antimicrobial peptide (CRAMP), eosinophil-derived neurotoxin (EDN) and granulysin; (2) nuclear form, including HMGB-1, HMGN1, IL-33, and IL-1α; (3) cytoplasmic, as heat shock proteins (HSP-60, -70, -90, and -96), S100 proteins, ATP and uric acid [17][18]. These intracellular proteins are generally released as inflammatory signal mediators and represent the first defense against infections, as well as during trauma and various metabolic, physical or chemical injuries [18][19][19,20]. Their release can attract additional inflammatory molecules, such as leukocytes, triggering a massive immune local response [20][21] and activating dendritic cells. Roh et al. [21][22] reported that alarmins and various damage-associated molecular patterns (DAMPS), such as HMGB1, S100 and HSP-70, play a key role in the pathogenesis of inflammatory diseases. Several studies have shown a key role for HMGB1, heat shock proteins and s100 proteins in AF physiopathology.
2. HMGB-1 and Human Studies
High-mobility group box-1 (HMGB1) protein, also called amphoterin, is an alarmin named for its high electrophoretic mobility. HMGB1, encoded by a gene located on chromosome 13q12, is a non-histone structural chromatin-binding protein with 215 amino acids and a molecular weight of 25 kDa. The protein contains two homologous proximal DNA-binding domains, called A-box (9–79 aa) and B-box (95–163 aa), and a C-terminal acidic tail (186–215 aa) with repeated glutamic and aspartic acid residues. Two nuclear localization signals (NLS1 and NLS2) and two nuclear export signals (NES) drive the translocation of HMGB1 from the nucleus to the cytoplasm. The localization of HMGB1 is pivotal for its function and plays a key role in acute and chronic inflammation, with effects in several diseases
[22][54]. HMGB1 is located in the nucleus, cytosol, or extracellular space, where it is passively released from lytic cell death or actively secreted by viable cells. Under physiological conditions, HMGB1 is anchored in the nucleus. When HMGB1 is released, it modulates cellular stress responses and inflammation
[23][55]. Several studies confirmed a key role of HMGB1 in AF (
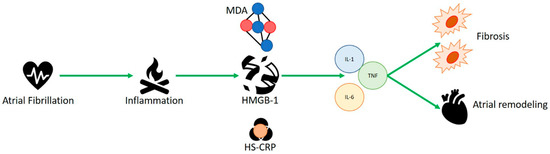
Figure 1). Qu et al. showed the higher incidence of postoperative AF in patients with rs2249825 polymorphism of HMGB1 protein receiving elective cardiac surgery (CABG). High levels of HMGB1 protein were related with genotype CG + GG versus genotype CC
[24][56]. Recent onset postoperative AF is one of the most common complications after cardiac surgery
[25][31]. The injury triggers a local inflammatory response that results in elevated serum concentrations of inflammatory biomarkers. This could represent a close correlation between oxidative stress and HMGB1; as the first increases, so does the second one. Such a concept would open possibilities for anti-AF therapies accurately targeting the inflammatory state underlying the pathology. Higher concentrations of tissue factor (TF) and HMGB1 protein were found in the left atrial appendage (LAA) tissue of 45 patients with AF and thrombosis receiving surgical valve replacement for rheumatic disease, compared with patients without thrombosis. Th
eis resultstudy showed a linear correlation of HMGB1 with TF, myeloid differentiation factor 88 (MyD88) and nuclear factor kB (NfkB)
[26][57]. It was also shown that high levels of oxidative stress proteins, such as malondialdehyde and hsCRP, are positively correlated with HMGB1 levels in AF patients. HMGB1 protein, in association with TF, plays a key role in the downstream regulation of MyD88/NfkB, suggesting that HMGB1 could cause thrombosis through the MyD88/NfkB pathway. The relationship between HMGB1 and thrombogenesis is carried out through the RAGE, TLR4 and MyD88 pathways, with activation of platelets (TLR-4, TLR-2), determining thrombosis in LAA. Another study suggests evidence for immune-mediated platelet activation (TRL-2, TRL-4 and HMGB1 protein) in the left atria of patients with AF, in which Toll-like receptor 2 and Toll-like receptor 4 were higher in persistent AF, which could suggest the hypothesis that the role of HMGB1 in atrial thrombogenesis in AF patients could be via TLR-2 and TRL-4
[27][32]. As is well known, the cardio-embolic risk caused by AF is the basis of the therapeutic pathway with the aim of reducing stroke incidence
[28][33]. While macroscopically, atrial noncompliance, atrial remodeling and fibrosis, and blood stasis may be causes, HMGB1 appears to independently control high thrombogenic activity in patients with AF and plays a key role in the thrombogenic process. This suggests a new role for HMGB1 in thrombogenesis in AF patients and new potential goals for future anticoagulant therapies. Increasing the serum concentration of HMGB1 was achieved in 86 patients with paroxysmal AF and persistent AF. The serum concentration of HMGB1 was higher in persistent AF patients
[29][58]. HMGB1 was discovered to have various functions within a molecular pathway. When it resides outside the cell, it is one of the most powerful signals of inflammation, mediated by the innate immune system; when it resides in the nucleus, HMGB1 binds and folds the DNA helix, with the purpose of protein formation and ensuring nuclear biochemical processes
[30]. In
thisour case, it appeared to be highly represented in patients with AF, correlating its concentration with that of MDA and HS-CRP, which represents another inflammatory protein. HMGB1 could be the amplifier of the inflammatory response with the recall of proinflammatory cytokines such as IL-1, tumor necrosis factor (TNF) and IL-6, which handle atrial remodeling and fibrosis, as well as in the onset of post-surgical AF. Moreover, exactly where the inflammatory mechanism is continuous and constant, as in permanent AF, it seems to occur in higher concentrations than in the paroxysmal AF condition, as an independent predictor of AF.
Figure 1. HMGB1 appears to be highly represented in patients with atrial fibrillation, correlating its concentration with malondialdehyde (MDA) and C-reactive protein (HS-CRP), which represents another inflammatory protein. HMBG1 could be amplifier of the inflammatory response with the recall of proinflammatory cytokines such as IL-1, tumor necrosis factor (TNF) and IL-6, which handle atrial remodeling, fibrosis and in the onset of post-surgical AF.
3. S100 Protein and Human Studies
RIn this re
searchersview, we also analyzed the role of the S100 proteins. Generally, S100s are calcium-binding proteins and modulators of various enzymatic activities, including differentiation, inflammation, proliferation, migration, apoptosis, Ca
2+ homeostasis, and energy metabolism
[31][32][59,60]. S100 proteins are a subfamily of EF-hand, calcium-binding proteins with integrated dimeric structure that can form higher-order oligomers
[33][61]. They represent some of the major second messenger transducers of intracellular Ca
2+ signals. They also play a significant role in the extracellular environment, which is pivotal in the innate immune response and activation of inflammation. Out of the 25 different S100 genes, only S100A7, S100A8, S100A9 and S100A12 have been identified as modulators of innate immunity. The family of S100 proteins is exceptionally large; to date, it appears that S100A4 and S100β are related to AF in the literature (
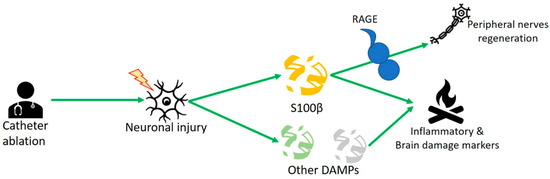
Figure 2). Scherschel et al. showed higher concentrations of S100β protein in AF patients undergoing catheter ablation. Neuronal injury by the intrinsic cardiac autonomic nervous system (ICSN) upon catheter treatment released damage-associated molecular pattern proteins (DAMPs) as S100β protein. In vitro studies dealing with murine intracardiac neurons showed that S100β decreased potential action and increased neuronal cell growth
[34][62]. This confirms its well-known role as an inflammatory biomarker. At the same time, the trophic role of S100β was also shown, which, released from glial cells through the RAGE receptor, would be able to regenerate peripheral cardiac nerves. Moreover, at 6 months after surgery, patients had fewer recurrences of AF. This would demonstrate a high protective role of the molecule in the onset of AF and could reveal its crucial role as a predictor marker of disease. Kato et al. showed that, in the atrial tissue of AF patients (paroxysmal AF, persistent AF) undergoing left atrial appendectomy during cardiac surgery, extension of atrial fibrosis was related with the amount of S100A4 protein
[35][27]. Another novel therapeutic approach to prevent atrial remodeling could be to evaluate S100A4 protein, known as fibroblast specific protein 1 (FSP1), related to endothelial–mesenchymal transition of endothelium atrial cells and with the amount of fibrosis and atrial size. It was demonstrated that high concentrations of this molecule positively correlated with fibrotic depositions in the left atrium and atrial size.
[30]. Scherschel et al.
[36][26] showed high levels of S100β protein in paroxysmal AF patients undergoing pulmonary vein isolation (PVI) with different techniques, either radiofrequency (RF) or cryoballoon (CB), confirming its role as a biomarker of neurological damage. A study of 243 patients showed that AF patients had high levels of cerebral injury-related circulation proteins, such as TAU protein, astrocyte-specific glial acidic fibrillary protein (GFAP), and growth differential factor 15 (GDF15), but no calcium-binding protein B S100β
[37][29]. Sramko et al.
[38][28] evaluated S100B concentrations in patients with atrial fibrillation after catheter ablation who underwent brain MRI before and after the procedure. They investigated whether detection of ablation-related brain damage could be improved by assessment of S100B protein, a biomarker of brain damage. Very high values were recorded immediately after the procedure in patients with permanent atrial fibrillation; moreover, they appeared to be directly related to atrial size. The analysis of these two studies led to the evaluation of S100β as a biomarker that is extremely sensitive to cardiac damage but not specific to brain damage. More importantly, this protein is an early marker of blood–brain barrier opening that may precede neuronal damage, possibly influencing future therapeutic strategies. Furthermore, high concentrations of S100β are indicators of recent brain damage and predictors of adverse pathology outcome or possible diagnostic means to differentiate extensive from minor and transient damage
[39][25].
Figure 2. S100β is released because of the neuronal injury of the intrinsic cardiac autonomic nervous system (ICSN), which releases other damage-associated molecular pattern proteins (DAMPs). At the same time, the trophic role of S100β is also shown, which, released from glial cells, through the RAGE receptor would be able to regenerate peripheral cardiac nerves.