Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Beatriz Tlatelpa Romero and Version 2 by Catherine Yang.
Diffuse parenchymal lung diseases (DPLD) or Interstitial lung diseases (ILD) are a heterogeneous group of lung conditions with common characteristics that can progress to fibrosis. Within this group of pneumonias, idiopathic pulmonary fibrosis (IPF) is considered the most common. This disease has no known cause, is devastating and has no cure.
- pulmonary surfactant
- phospholipids
- lung fibrosis
1. Introduction
Diffuse parenchymal lung diseases (DPLD) or Interstitial lung diseases (ILD) are a heterogeneous group of lung conditions with common characteristics that can progress to fibrosis. Within this group of pneumonias, idiopathic pulmonary fibrosis (IPF) is considered the most common. This disease has no known cause, is devastating and has no cure. Chronic lesion of alveolar type II (ATII) cells represents a key mechanism for the development of IPF. ATII cells are specialized in the biosynthesis and secretion of pulmonary surfactant (PS), a lipid-protein complex that reduces surface tension and minimizes breathing effort. Some differences in PS composition have been reported between patients with idiopathic pulmonary disease and healthy individuals, especially regarding some specific proteins in the PS; however, few reports have been conducted on the lipid components. This revientryw focuses on the mechanisms by which PLs could be involved in the development of the fibroproliferative response.
2. ILD of Know Cause: Occupational Exposure
1.1. Asbestosis
Asbestosis is pneumoconiosis caused by inhaling asbestos fibers, which elicit potent inflammatory responses in the respiratory tract [1][55]. It has been shown that asbestos can cause lipid peroxidation and trigger genomic alterations in pulmonary cells [2][56]. Furthermore, a decrease in phospholipase C (PLC) activity has been described, which directly impacts the biosynthesis of lipids like phosphatidylinositol, phosphatidylglycerol and phosphatidylserine, affecting macrophage activity and PS availability [3][57]. In asbestosis, alterations in the alveolar activity of CC-16 (specific proteins of clear cells), SP-A, and phospholipase A2 (PLA2) have been reported. [4][58]. The activity of PLA2 secreted in alveolar fluids and the effect of CC-16 on platelet growth factor-induced chemotaxis of human fibroblast [5][59]. SP-A is associated with lung diseases such as IPF and alveolar proteinosis [6][60], while increased PLA2 has been implicated in inflammatory lung disease pathologies such as respiratory distress syndrome, asthma and alveolar proteinosis [5][59]. A consequence of asbestos parenchymal damage is pulmonary fibrosis. The molecular mechanisms by which it could occur are primarily by the death of ATII cells mediated by mitochondrial and p53 death pathways resulting from the production of iron-derived reactive oxygen species (ROS) and DNA damage [7][61]. In the development of pulmonary fibrosis caused by asbestos, the death of ATII and the decrease in macrophage activity could be involved (Figure 1).
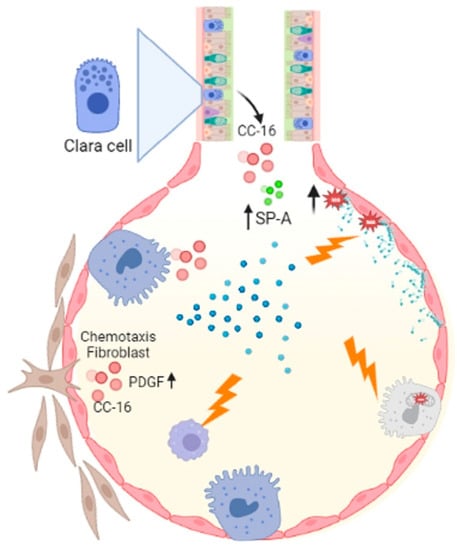
Figure 1. Asbestosis influences macrophage activity which is important for eliminating cellular debris, and acts on ATII cells by activating the mitochondrial and p53 death pathways. Asbestos catalyzes lipid peroxidation by decreasing PLC, which decreases phosphatidylinositol, phosphatidylglycerol, and phosphatidylserine synthesis. Another important event is the alteration of CC-16 with fibroblast chemotaxis activity, increased Sp-A and PLA2. Figure created in BioRender.com.
1.2. Silicosis
Occupational exposure to crystalline-free silica SiO2 dust causes silicon nodules and diffuse pulmonary fibrosis. Lung function is severely damaged by silicosis fibrosis, whose degree increases over time [8][62]. The alveoli of lungs affected by acute silicosis are lined by hypertrophic ATII, which produce proteinaceous materials, and PS is produced, resulting in protein-filled alveoli [9][10][63,64]. A high level of PS components in the lungs is a result of exposure to silica dust [11][65]. Disaturated phosphatidylcholine and SP-A levels increased in intracellular and extracellular PS compartments following silica exposure in rats [12][66]. A high concentration of silica reduces the number of alveolar macrophages, which are responsible for decreased DPPC liposomes during PS metabolism [13][67] (Figure 2).
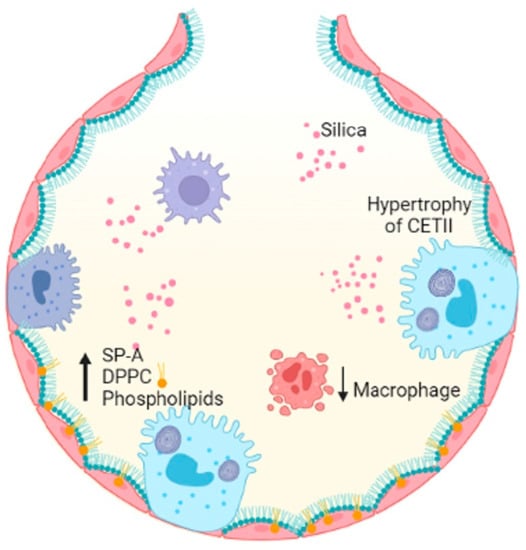
Figure 2. Silica entering the alveolar space decreases the number of macrophages, induces hypertrophy of ATII cells and results in increased SP-A, DPPC and PLs, damaging alveolar homeostasis. Figure created in BioRender.com.
3. ILD of Know Cause: Related to Treatment
3.1. Radiation
Radiation therapy (also called radiotherapy) kills cancer cells and shrinks tumors by administering high doses of radiation. However, the use of radiotherapy can cause acute or chronic pneumonia [14][68]. The inflammatory process of acute pneumonia involves the formation of intra-alveolar and septal edema, as well as the formation of epithelial and endothelial desquamations. During acute pneumonia, ATII cells play a critical role in increasing microvascular permeability. This hypothesis is supported by the findings that ATII is the primary cellular response to lung injury. Then, a secondary response of ATII cell hyperplasia occurs as a result of the plasma protein overload on lymphatic and other drainage mechanisms. Consequently, an excess of PS results, causing a fall in compliance, abnormal gas exchange values, and even respiratory failure. As a result of the inflammatory process, chronic fibrosis can develop [15][69]. Other studies reveal that radiation causes apoptosis of epithelial cells, as well as a large amount of oxidative damage and release of fibrotic cytokines [16][70]. Oxidative damage affects lipid metabolism, increases inflammatory cytokines such as interferon-γ (INF-γ) and tumor necrosis factor-α (TNF-α) by macrophages [17][71]. Radiation increases glucose catabolism by increasing the levels of glycerophosphate as a lipid precursor [18][72], increases the expression of lipoproteins lipase and fatty acid binding protein (FABP4) and triacylglycerides, all resulting in lipid accumulation [19][73] (Figure 3).
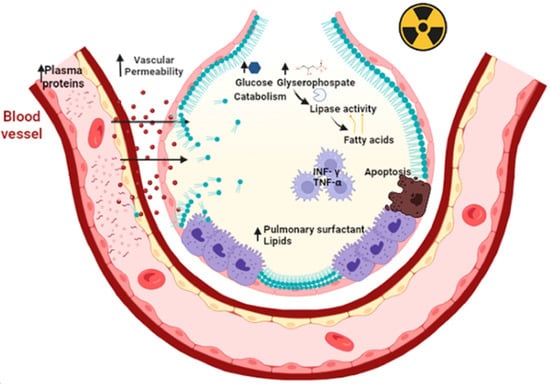
Figure 3. Primary radiation increases microvascular permeability and plasma proteins that overwhelm the drainage mechanisms. This elicits a secondary response to hyperplasia of type II epithelial cells. In addition, there is an accumulation of pulmonary surfactant within the alveolus that triggers apoptosis of type II epithelial cells, oxidative damage and the release of fibrotic cytokines such as TNF-Υ and TNF-α. Primary radiation increases the catabolism of lipoprotein lipases due to increased glucose and glycerophosphate. This results in the release of fatty acids leading to decreased distensibility and respiratory failure. Figure created in BioRender.com.
3.2. Amiodarone
Amiodarone is a highly toxic antiarrhythmic drug that contains iodinated benzofuran, which causes fibrosis in the lungs [20][74]. Several studies have demonstrated that the accumulation of multilamellar bodies in the cytoplasm is the result of reduced degradation of PLs since amiodarone inhibits lysosomal phospholipases A1 and A2 [21][22][75,76]. Furthermore, amiodarone and the metabolite desethylamiodarone had a high capacity to bind to DPPC and prevent the reuptake of LBs [22][76]. Therefore, phospholipid accumulation and oxidative stress may contribute to pulmonary fibrosis through amiodarone exposure, so new therapies are aimed at modulating lipid homeostasis (Figure 4).
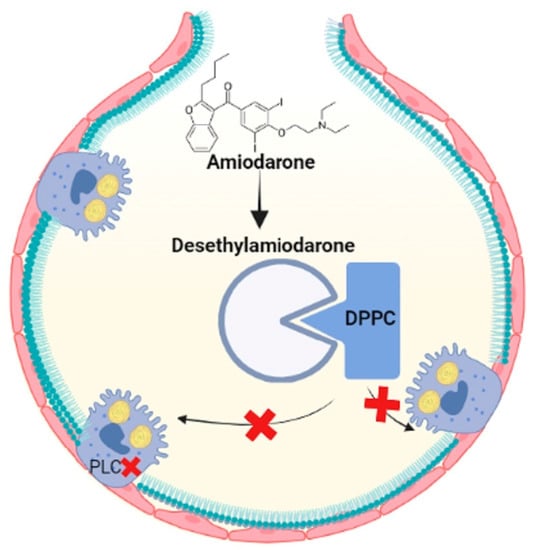
Figure 4. Amiodarone is metabolized to desethylamiodarone, which binds to DPPC, prevents it from being taken up by ATII cells and inhibits phospholipases and increases lipids in the alveolar space. Figure created in BioRender.com.