Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 4 by Beatrix Zheng and Version 3 by Francesca Oppedisano.
Mitochondrial aldehyde dehydrogenase 2 (ALDH2) is a cardioprotective enzyme that catalyzes the bio-activation of GTN to NO. Nitrate tolerance is accompanied by an increase in oxidative stress, endothelial dysfunction, and sympathetic activation, as well as a loss of the catalytic activity of ALDH2 itself. On the basis of current knowledge, nitrate intake in the diet would guarantee a concentration of NO such as to avoid (or at least reduce) treatment with GTN and the consequent onset of nitrate tolerance in the course of cardiovascular diseases, so as not to make necessary the increase in GTN concentrations and the possible inhibition/alteration of ALDH2, which aggravates the problem of a positive feedback mechanism.
- aldehyde dehydrogenase 2 (ALDH2)
- nitrate tolerance
- nitric oxide (NO)
1. Vascular Endothelium and Endothelial Dysfunction
The endothelium is a cell monolayer composed of approximately 1013 endothelial cells (ECs). This layer covers the inner surface of the blood vessels, lymphatic vessels, and heart. In relation to the mesenchymal derivation, the endothelium is the largest tissue in the body, with a total weight of around 1.0–1.8 kg. Furthermore, its cells represent about 1.5% of total body mass [1]. Until the early 1980s, the endothelium of the vascular tree was believed to play a passive role in forming the shell of the vascular shaft, dealing only with selective permeability to water and electrolytes. Today, it is known that the endothelium carries out numerous functions, including regulation of the tone and the structure of the vessels [2]; regulation of vasal permeability [3]; angiogenesis [4]; hemostasis checks [5]; control of inflammation and recruitment of neutrophils [6]; and endocrine–metabolic functions [7]. It is possible to define the endothelium as an endocrine, paracrine, and autocrine organ, which is capable of releasing a wide variety of substances in the blood and interstitial space (including vasoactive compounds, growth factors, inflammation mediators, adhesion molecules, hemostatic system proteins, and extracellular molecules). These substances can act remotely (via endocrine activity), on nearby cells (via paracrine activity), or on the cell that produced them (thereby demonstrating an autocrine activity); it is these activities that are responsible for the balance maintained by the endothelium [8]. In particular, blood flow is regulated through the secretion and absorption of vasoactive substances by the endothelium, which act in a paracrine fashion to shrink and dilate specific vascular beds [9][10]. When the endothelium is working normally, all the appointed functions are carried out adequately, which consequently also involves a proper immune response. On the other hand, endothelial dysfunction characterized by reduced vasodilation—which is a pro-inflammatory state and when active pro-thrombic properties are present—is associated with most forms of cardiovascular diseases, such as coronary heart disease, hypertension, diabetes, chronic kidney failure, peripheral vascular disease, and severe viral infections [11][12]. Endothelial dysfunction is involved in other regions [13][14] and also causes considerable damage to the nervous system [15][16][17][18]. Vascular tone is defined by the balance between the degree of constriction of the blood vessel and its maximum dilation; in addition, it is modulated by the release of relaxing as well as constrictive factors derived from the endothelium. In fact, ECs physiologically synthesize and release several relaxing factors derived from the endothelium—including vasodilator prostaglandins, endothelium-dependent hyperpolarization factors, and NO, but also contraction factors such as thromboxane A2, endothelin, angiotensin II, superoxide, and the correct balance between the production of vasodilators and vasoconstrictors (which ensures proper maintenance of vascular tone) [1]. NO is a soluble gas that demonstrates important vaso-relaxant protective functions and is regulated by nitric oxide endothelial synthase (e-NOS), which is an enzymatic isoform constitutively expressed in ECs. In particular, this enzyme catalyzes the conversion of L-arginine to L-citrulline and NO. When NO is synthesized, it spreads into smooth vascular muscle cells, stimulating soluble guanylate cyclase and the increasing of cyclic guanosine monophosphate (which is an NO effector that promotes vasodilation) [19]. Reactive oxygen species (ROS) are reactive oxygen intermediates that form physiologically as byproducts of cell metabolism. When present at physiological concentrations, ROS are very useful for cellular homeostasis, acting as second messengers in the transduction of cellular signals and predisposing toxicity reactions against bacterial infections [20]. On the other hand, when ROS levels exceed the antioxidant capacities of the cell or when the antioxidant enzymes have reduced activity, the onset of oxidative stress occurs. This condition is extremely dangerous, as ROS can react with major biological macromolecules and alter them accordingly [21][22]. Cell membranes are particularly susceptible to oxidative damage caused by ROS and can encounter “lipid peroxidation”, a process in which ROS remove electrons from lipids and damage phospholipids. This alteration can also lead the cell to apoptotic death [23][24]. ROS accumulation is involved in the onset of several diseases, including cancer, as well as many metabolic diseases such as diabetes and obesity, neurodegenerative disorders, lung diseases, and kidney diseases [25][26][27][28], among others. It has recently been shown that there is a close correlation between accumulation of ROS and increased inflammation and endothelial dysfunction [29][30][31]. For example, the reduction in bioavailability of NO can occur not only for decreased e-NOS protein expression, but also as a result of an increased level of ROS and, especially, of superoxide anions (O•−), which are responsible for the formation of peroxynitrite (ONOO−). The latter promotes protein nutrition by contributing to endothelial dysfunction and cellular death [32][33]. In most instances, the physiological antioxidant mechanisms of the human body are able to neutralize ROS [34]. However, in their absence endothelium lesions, relative dysfunctions, and alterations in the content of NO can occur [35][36]. Endothelial dysfunction—which is related to the production and accumulation of ROS, as well as reactive nitrogen species (RNS)—is particularly involved in some pathologies, such as hypertension, hyperlipidemia, atherosclerosis, and diabetes mellitus, which all have connections with vascular damage as a common denominator [37]; in fact, endothelial dysfunction can be considered an early marker of cardiovascular events [38][39]. In smooth muscles, the listed pathologies or bad habits—including smoking and alcohol intake—can lead to activation of the enzyme NADPH oxidase, which is responsible for the desensitization of soluble guanyl cyclase and the breakdown of cyclic guanosine monophosphate [40]. The close correlation between endothelial damage and cardiovascular risk is also associated with decreased NO bioavailability, as well as impaired activity of endothelial NO synthase [41][42]. Today, it is known that endothelial dysfunction also involves the alteration of ALDH2 expression, thereby resulting in the alteration of the oxidative state and the onset of inflammation [43]. Due to the fact that, as already described, ALDH2 protects endothelial cells from oxidative stress, these cells are thus able to turn their physiological function on or off [44][45]. A representation of this double control of the endothelium regarding ALDH2 is shown in Figure 1.
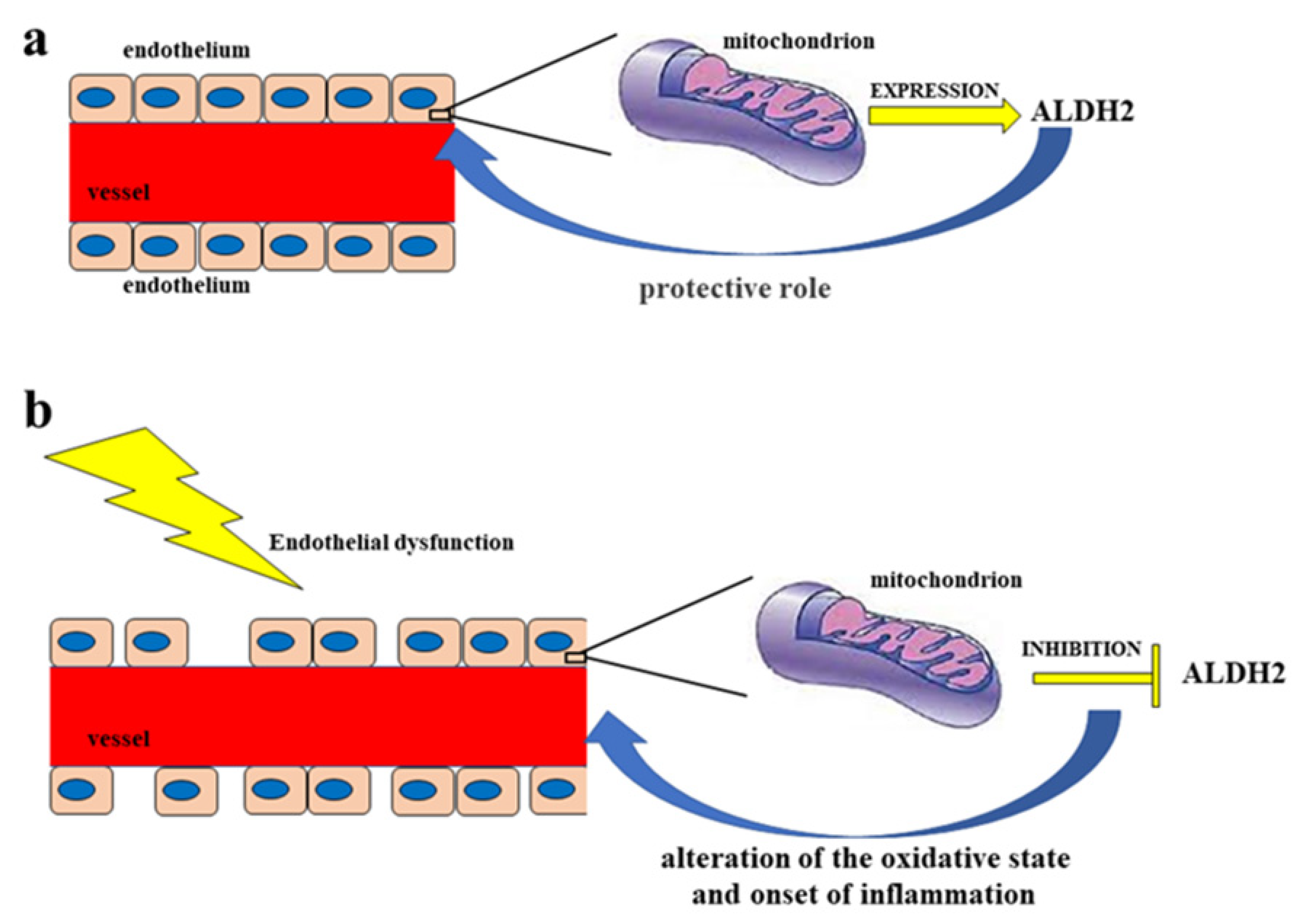
Figure 1. Double control of the endothelium regarding ALDH2. Diagram (a) represents a functioning endothelium with a proper mitochondrial expression of the enzyme ALDH2, which has a protective action on the endothelium itself. On the other hand, in diagram (b) the dysfunctional endothelium determines the inhibition of the mitochondrial synthesis of ALDH2, with a consequent negative effect on the endothelium, which is undergoing an alteration of the oxidative state and the onset of inflammation.
Nitrates, Cardiovascular Diseases, and Tolerance
Reduced bioavailability of the NO signaling molecule due to its reduced synthesis or excessive consumption has been associated with the occurrence of cardiovascular diseases. For this reason, its restoration guarantees a mechanism that has a positive effect on these pathologies [46]. In addition, it is important to act on the reduced availability of substrates and cofactors, on the generation of ROS, and on the oxidation of NOS, which are factors that disturb the signaling NO and accentuate the development and pathogenesis of cardiovascular diseases [47]. A highly interesting source of indirect syntheses of NOs is provided by the inorganic anions nitrate (NO3−) and nitrite (NO2−). In fact, nitrates and nitrites are physiologically transformed in the blood and tissues to form NO and other oxides of bioactive nitrogen; moreover, they should be considered a reserve of NO. Nitrates are transformed into nitrites in the gastrointestinal tract. This is achieved through the reducing power of some intestinal bacteria. On the other hand, nitrites are reduced to NO in the oral cavity and stomach, where the pH is particularly acidic. In this way, the nitrate–nitrite–NO path becomes complete [48]. The trinomial nitrate–nitrite–NO is fundamental in restoring the health of the cardiovascular system and generating NO-similar protective effects. This is in addition to the fact that it is widely used as a treatment in myocardial infarction, hypertension, and peripheral artery diseases [49]. The hypothesis that nitrates could produce an effect similar to that of NO was confirmed in 2006 by a research group who demonstrated that the administration of sodium nitrate for 3 days was able to reduce blood pressure in young, healthy individuals [50]. Subsequently, these protective effects of nitrates were confirmed in animal and human models of hypertension, oxidative stress, ischemia-reperfusion, and tolerance to hypoxia [51]. At the same time, the effects of nitrites on cardiovascular function were also examined [52], and with the same protective effect as NO, the nitrate–nitrite–NO pathway has become a therapeutic opportunity to be exploited in the event of cardiovascular disorders [53]. For example, Stokes et al. showed that dietary nitrite intake in mice with endothelial dysfunction (which was induced by dietary hypercholesterolemia) was able to preserve endothelial function, inhibit microvascular inflammation, and reduce increased expression of the C-reactive protein [54]. Another group found that nitrite intake reduced arterial stiffening and oxidative stress in a mouse model of endothelial dysfunction related to aging [55]. Moreover, sodium nitrate ingestion was able to mitigate endothelial function on ischemia-induced effects [56], reduce blood pressure [57], and attenuate cardiac hypertrophy and fibrosis in chronic models of hypertension [58][59]. Webb et al. showed, in an ex vivo mouse model of myocardial infarction, that nitrite could reduce the damaged area via a mechanism that involved the formation of NO [60]. Having said this, not only the heart was affected, but also other organs including the brain [61], kidneys [62], liver [63], and the hind limbs [64]. The mechanism of cytoprotective action carried out by nitrite appears to be the inhibition of mitochondrial respiration, with consequent reduction in the formation of ROS [65][66]. The encouraging effects of nitrates and nitrites as substrates for an in vivo formation of NO and its related oxides of nitrogen bioactives have stimulated the study of these clinical compounds in animal models; in particular, the beneficial effects justified considering these molecules as an opportunity for the development of drugs against cardiovascular and metabolic diseases. For this reason, phase II studies have been initiated on a model of acute myocardial infarction [67]. The use of organic nitrite was replaced by an organic nitrate, GTN, which was easier to administer and had a longer duration of action. GTN has been used in the treatment of angina pectoris and heart failure for over 150 years. The use of GTN results in the release of NO, the activation of guanyl cyclase, and the relaxation of blood vessels [68]. Although the protective effects of organic nitrates on cardiovascular dysfunction are undisputed, it is also known that chronic therapy with these compounds leads to long-term multifactorial effects that can be summarized as the development of a tolerance to nitrates; the onset of profound changes in vascular homeostasis with oxidative stress; and a suspicion that organic nitrate therapy may be associated with an increase in coronary events and neurohumoral adaptations [69]. Nitrate tolerance is defined as the loss of the hemodynamic effects of organic nitrates and the need for higher dosages in order to maintain the same effects. It is the consequence of a series of phenomena, both vascular and extravascular, which can generate important clinical implications [70]. In the development of nitrate tolerance, the role of mitochondrial oxidative stress was described in a mouse model that had a heterozygous deletion of the enzyme manganese superoxide dismutase (MnSOD), which is the mitochondrial isoform of this enzyme. In addition, ROS has been shown to be responsible for disrupting cross-talk to the cytoplasm and mitochondria [71]. Interestingly, nitrates are responsible for inhibiting ALDH2, which is responsible for decreasing or canceling the protective mechanism against oxidative stress [72]. GTN can potently and rapidly inactivate ALDH2, which is an even earlier effect of nitrate tolerance. It was shown, for example, that in knockout mice for ALDH2, nitrate tolerance occurs more easily [73]. The severity of the tolerance is responsible for several effects, as is represented in Figure 2. Due to the discomfort of long-term administration of nitrates, it is essential to select a storage form of NO that may have an important therapeutic potential and advantages over organic nitrates.

Figure 2. The severity of nitrate tolerance and its effects.
2. The Role of ALDH2
ALDH2 is considered a cardioprotective enzyme [74][75][76] that is capable of both preventing the onset of ischemic damage (during myocardial infarction) and also in reducing the infarct area [74]. In humans, the aldehyde dehydrogenase (ALDH) superfamily consists of nineteen NAD(P)+-dependent isozymes. These catalyze the oxidation of both exogenous (such as alcohol) and endogenous (such as lipids and amino acids) aldehydes into carboxylic acids [77][78][79][80][81][82]. This protects the cells from damage caused by active aldehydes and plays an important role in ROS elimination. In fact, high concentrations of non-metabolized aldehydes cause enzyme inactivation, DNA damage, and cell death [79]. Among these, ALDH2 is present in various organs, including the heart; in particular, it is localized at the level of mitochondria, organelles that are important for ROS and reactive aldehyde generation [83][84]. It is a tetrameric allosteric enzyme involved in the metabolism of ethanol. The latter is first converted to acetaldehyde in a reaction catalyzed by alcohol dehydrogenase (ADH) and then forms acetic acid in a reaction catalyzed by ALDH2 [83]. In addition to its dehydrogenase activity, ALDH2 also possesses esterase and reductase activity. Furthermore, its role in GTN-induced vasodilation is linked to its reductase activity [85]. The mechanism by which ALDH2 catalyzes the bioactivation of GTN to NO has been studied in recent years by various research groups. In particular, Lang et al. (2012), by means of crystallography and mass spectrometry studies conducted on wild-type ALDH2 and on the triple mutant of the protein with reduced denitration activity (E268Q/C301S/C303S), tried to elucidate this mechanism. Indeed, it has been established that the denitration of GTN begins with the nucleophilic attack of Cys-302, present in the catalytic site of the enzyme, on a terminal nitrogen of GTN. This results in the formation of a thionitrate adduct as the first reaction intermediate and the release of 1,2-glyceryl dinitrate (1,2-GDN). At this point, the intermediate can undergo nucleophilic attack by the flanking cysteines, Cys-301 or Cys-303, with the formation of nitrites and a disulfide bond in the active site of the enzyme, which would lead to the reversible inhibition of ALDH2. The released nitrite would subsequently be reduced to NO, or ALDH2 could be irreversibly inhibited with the formation of sulfinic acid in the active site, mediated by the presence of Glu-268. This pathway could play a fundamental role in the development of nitrate tolerance. A third pathway leads to the direct production of NO and the reversible inhibition of ALDH2 [86].
2.1. Oxidative Stress, Toxic Aldehydes, and ALDH2
Certain studies conducted in vivo by performing pretreatments with nitroglycerin for 8 days (as well as in vitro studies on isolated rat thoracic aortas and on human umbilical vein endothelial cells (HUVECs) treated with nitroglycerin) have shown that, during the nitrate tolerance process, an increased ROS formation reduces ALDH2 activity. This phenomenon results in a reduction in the release of the endogenous calcitonin gene-related peptide (CGRP) [87]. Hence, chronic nitroglycerin treatment results in an increase in mitochondrial and vascular oxidative stress, as well as a reduction in ALDH2 activity in the aorta and cardiac mitochondria at the same time [88]. In fact, during heart failure (HF), ROS and oxidative stress play a fundamental role both in the phase of myocardial remodeling and in the condition of overt HF. It is known that the intracellular ROS sources are due to the activities of NADPH oxidase (NOX), an enzyme that with its two isoforms NOX1 and NOX4 generates superoxide, clinically associated with atherosclerosis and therefore cardiovascular disorders [89], xanthine oxidase, and nitric oxide synthase [90]. This is in addition to the activity of an enzyme localized in the external mitochondrial membrane, monoamine oxidase (MAO)—of which there are two isoforms, MAO-A and -B. MAO-catalyzed oxidative deamination reactions produce hydrogen peroxide and aldehydes, which can be eliminated by ALDH2 [90]. A reduction (or an inhibition) in ALDH2 activity leads to the accumulation of toxic aldehydes generated by MAO, thereby causing mitochondrial dysfunction and consequent myocardial failure. This supports the fundamental role of ALDH2 in eliminating toxic aldehydes in the myocardium in order to protect the heart from oxidative stress [90]. It is reported that in the vascular endothelium, the loss of ALDH2 activity leads to endothelial dysfunction, as there is an increase in ROS levels, an accumulation of 4-hydroxy-2-nonenal (4-HNE) protein adducts, and a loss of mitochondrial bioenergetic functions. Therefore, senescence of the endothelium with loss of regenerative capacity can be a defensive response to the damage caused by the accumulation of toxic aldehydes, thus leading to a loss of function in the vascular system [44]. Furthermore, in a study conducted on cell model RAW264.7 that was treated with oxidized low-density lipoprotein (ox-LDL), it was determined that ALDH2 activation reduces ox-LDL-induced 4-HNE production. Therefore, oxidative stress in atherosclerosis could be reduced through the inhibition of activation of the NLRP3 inflammasome by ALDH2, making it a potential target for anti-inflammatory therapies [91][92].
2.2. ALDH2 and Ischemia-Reperfusion Injury (IRI)
It is known that ALDH2 has a cardioprotective function, which also limits ischemia-reperfusion injury (IRI) [83][84][93][94]. This condition arises as a consequence of reperfusion, which is a necessary treatment to reduce the magnitude of myocardial infarction, as well as the direct manifestation of coronary artery disease (CAD). IRI is also generated following cardiac arrest due to open-heart surgery with cardiopulmonary bypass (CPB), in which there is an increase in the synthesis of superoxide, ROS, and aldehydes. ALDH2 activation has been reported to reduce ischemic cardiac damage. Furthermore, there are various mechanisms underlying the cardioprotective effects of ALDH2. Among these, ALDH2 acts downstream of protein kinase C type ε (εPKC). In particular, it reduces the production of reactive aldehydes, such as 4-HNE, that are produced during the peroxidation lipid process. The cardioprotective effect of ALDH2, through its action on reactive aldehydes, has positive consequences in regard to ROS production, mitochondrial kATP channel regulation, and mitochondrial permeability transition pore (MPTP) openings [83][84][95][96][97]. In addition, the apoptotic process in cardiomyocytes also increases during IRI following an increase in ROS production. ALDH2, by reducing ROS production, reduces the activation of the JNK signaling pathway and the expression of c-Jun [83][84]. The autophagic process also increases during IR. Furthermore, ALDH2 protects cardiomyocytes by activating the LKB1/AMPK/mTOR pathway during the ischemic phase, as well as by acting on the PTEN/Akt/mTOR pathway during the reperfusion process, which in turn reduces excessive autophagy. Another cardioprotective mechanism of ALDH2 during IR is carried out through the reduction in the formation of reactive carbonyl species (RCS) that causes the carbonylation of proteins, with a consequent loss of activity [83]. In addition to this, in patients with CAD, ALDH2 plays a fundamental role in nitroglycerin metabolism, specifically for its bioconversion into NO in order to obtain a vasodilating effect [83]. It is known that after myocardial infarction, the co-administration of nitroglycerin and Alda-1—an activator of ALDH2—has positive effects on the metabolism of reactive aldehyde adducts by reducing the cardiac dysfunction generated by the use of nitroglycerin [85]. Indeed, in an animal model of IR, it has been shown that Alda-1, when administered before prolonged treatment with GTN, protects against heart damage from GTN-induced ALDH2 inactivation [98].
2.3. ALDH2 Polymorphism
In the liver, alcohol that is consumed is metabolized into acetaldehyde in a reaction catalyzed by ADH. In the subsequent reaction catalyzed by ALDH, acetaldehyde is converted to acetate. The latter leaves the liver and is metabolized in the heart and muscles. As an aside, acetaldehyde is a very toxic intermediate. Moreover, there are two isoforms of ALDH; one is cytosolic and encoded by the ALDH1 gene and the other is mitochondrial and encoded by the ALDH2 gene. The mitochondrial isoform is very important to this process due to its great affinity for acetaldehyde (Km = 0.20 μM) [99][100]. The polymorphism of the ALDH2 gene is linked to the role that the enzyme plays in the oxidation of alcohol. In fact, in exon 12, there is a single nucleotide polymorphism (SNP) that determines the substitution, at residue 504, of glutamic acid with lysine. The first condition corresponds to the ALDH2*1 allele, while the 504 Lys allele is referred to as ALDH2*2 and gives rise to a less active isozyme with a reduced ability to eliminate acetaldehyde. In regard to the people carrying this allele, the accumulation of acetaldehyde after alcohol consumption generates flushing, nausea, or vomiting, thereby increasing the risk of developing alcohol-related diseases [99]. The two isozymes are present in the Caucasian population, while about 30–50% of the East Asian population inherited the mutant ALDH2*2 allele [101]. ALDH2 is considered the most important enzyme for GTN bioactivation, as it activates GTN at clinically relevant plasma concentrations, i.e., below 1 μM. Despite this, forearm blood flow (FBF) responses to a brachial artery infusion of nitroglycerin in subjects with and without ALDH2 Glu504Lys polymorphism showed that ALDH2 is not the only enzyme responsible for the bioactivation of nitroglycerin at therapeutically relevant or higher concentrations. Studies of ALDH2 Glu504Lys polymorphism are important, as Glu504Lys is a common genetic variant that greatly reduces ALDH2 activity [102]. Taking into account that the enzymatic activities of the wild type (Glu504, encoded by ALDH2*1) and mutant (Lys504, encoded by ALDH2*2) proteins vary as a function of the functional genetic polymorphism of the protein [103][104], Miura et al. conducted a study on the vasodilator effect of GTN sublingual tablets administered to three different Japanese genotypic groups (ALDH2*1/*1, ALDH2*1/*2, and ALDH2*2/*2). The results of in vivo vasodilation determined by sublingual GTN did not show differences between the genotypes, even with respect to the degree of vasodilation. Having said this, the enzymatic activity of ALDH2 was different between the various groups studied, thus indicating the presence of other pathways aside from that of ALDH2 for the purposes of nitroglycerin bioactivation [103]. In addition, a study was conducted on patients with coronary spastic angina (CSA) in order to determine any differences in the response of nitroglycerin-mediated dilation (NMD) and flow-mediated dilation (FMD) between the wild type ALDH2*1/*1 and mutant ALDH2*2 (E487K point mutation). The results obtained by Mizuno et al. demonstrated that all patients reported comparable endothelial dysfunction as well as nitrate tolerance following continued GTN administration for 48 h. In patients with the ALDH2*2 mutation, however, tolerance was more severe, with a lower response to GTN at baseline [105]. Thus, treatment of coronary heart disease with GTN in ALDH2*2 subjects is clinically ineffective. Therefore, a reduced ALDH2 activity determines an inefficient elimination of reactive aldehydes with a consequent increase in cytotoxicity and oxidative stress [106]. In ALDH2*2 mutant mice, the administration of empagliflozin (EMP), a sodium–glucose cotransporter (SGLT) 2 inhibitor, has been shown to reduce the onset of diabetic cardiomyopathy via limiting the formation of 4HNE protein adducts, due to the condition of hyperglycemia. This improvement in cardiomyopathy occurs despite the mutant mice possessing low ALDH2 activity. The action of EMP is also reported in diabetic patients with the same mutation [107]. A study conducted in ALDH2*2 mutant mice demonstrated that coenzyme Q10—in addition to improving mitochondrial oxidative stress and preserving bioenergetics—is effective in protecting against attacks of atrial fibrillation (AF). The multi-omics studies conducted have made it possible to establish that coenzyme Q10 could be administered in humans who are characterized by the ALDH2*2 genotype in order to ensure protection from AF attacks [108].
References
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114.
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411.
- Jamwal, S.; Sharma, S. Vascular endothelium dysfunction: A conservative target in metabolic disorders. Inflamm. Res. 2018, 67, 391–405.
- Tang, X.; Wang, J.J.; Wang, J.; Abboud, H.E.; Chen, Y.; Zhang, S.X. Endothelium-specific deletion of Nox4 delays retinal vascular development and mitigates pathological angiogenesis. Angiogenesis 2021, 24, 363–377.
- Lichota, A.; Szewczyk, E.M.; Gwozdzinski, K. Factors Affecting the Formation and Treatment of Thrombosis by Natural and Synthetic Compounds. Int. J. Mol. Sci. 2020, 21, 7975.
- Dehghani, T.; Panitch, A. Endothelial cells, neutrophils and platelets: Getting to the bottom of an inflammatory triangle. Open Biol. 2020, 10, 200161.
- Suganya, N.; Bhakkiyalakshmi, E.; Sarada, D.V.; Ramkumar, K.M. Reversibility of endothelial dysfunction in diabetes: Role of polyphenols. Br. J. Nutr. 2016, 116, 223–246.
- Triggle, C.R.; Ding, H.; Marei, I.; Anderson, T.J.; Hollenberg, M.D. Why the endothelium? The endothelium as a target to reduce diabetes-associated vascular disease. Can. J. Physiol. Pharmacol. 2020, 98, 415–430.
- Xu, S. Therapeutic potential of blood flow mimetic compounds in preventing endothelial dysfunction and atherosclerosis. Pharmacol. Res. 2020, 155, 104737.
- Morales-Acuna, F.; Ochoa, L.; Valencia, C.; Gurovich, A.N. Characterization of blood flow patterns and endothelial shear stress during flow-mediated dilation. Clin. Physiol. Funct. Imaging 2019, 39, 240–245.
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967.
- Domingueti, C.P.; Dusse, L.M.; Carvalho, M.D.; de Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Complicat. 2016, 30, 738–745.
- Maiuolo, J.; Mollace, R.; Gliozzi, M.; Musolino, V.; Carresi, C.; Paone, S.; Scicchitano, M.; Macrì, R.; Nucera, S.; Bosco, F.; et al. The Contribution of Endothelial Dysfunction in Systemic Injury Subsequent to SARS-Cov-2 Infection. Int. J. Mol. Sci. 2020, 21, 9309.
- Maiuolo, J.; Carresi, C.; Gliozzi, M.; Mollace, R.; Scarano, F.; Scicchitano, M.; Macrì, R.; Nucera, S.; Bosco, F.; Oppedisano, F.; et al. The Contribution of Gut Microbiota and Endothelial Dysfunction in the Development of Arterial Hypertension in Animal Models and in Humans. Int. J. Mol. Sci. 2022, 23, 3698.
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Scicchitano, M.; Carresi, C.; Scarano, F.; Bosco, F.; Nucera, S.; Ruga, S.; Zito, M.C.; et al. The "Frail" Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections. Int. J. Mol. Sci. 2018, 19, 2693.
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Macrì, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Ruga, S.; et al. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. Int. J. Mol. Sci. 2019, 20, 3022.
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Scarano, F.; Nucera, S.; Scicchitano, M.; Bosco, F.; Ruga, S.; Zito, M.C.; et al. From Metabolic Syndrome to Neurological Diseases: Role of Autophagy. Front. Cell Dev. Biol. 2021, 9, 651021.
- Maiuolo, J.; Muscoli, C.; Gliozzi, M.; Musolino, V.; Carresi, C.; Paone, S.; Ilari, S.; Mollace, R.; Palma, E.; Mollace, V. Endothelial Dysfunction and Extra-Articular Neurological Manifestations in Rheumatoid Arthritis. Biomolecules 2021, 11, 81.
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiol. 2017, 219, 22–96.
- Abdul-Muneer, P.M.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2015, 51, 966–979.
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42.
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183.
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843.
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090.
- Khan, T.A.; Hassan, I.; Ahmad, A.; Perveen, A.; Aman, S.; Quddusi, S.; Alhazza, I.M.; Ashraf, G.M.; Aliev, G. Recent Updates on the Dynamic Association Between Oxidative Stress and Neurodegenerative Disorders. CNS Neurolrgets 2016, 15, 310–320.
- Saha, S.K.; Lee, S.B.; Won, J.; Choi, H.Y.; Kim, K.; Yang, G.M.; Dayem, A.A.; Cho, S.G. Correlation between Oxidative Stress, Nutrition, and Cancer Initiation. Int. J. Mol. Sci. 2017, 18, 1544.
- Lin, Y.C.; Chang, Y.H.; Yang, S.Y.; Wu, K.D.; Chu, T.S. Update of pathophysiology and management of diabetic kidney disease. J. Formos. Med. Assoc. 2018, 117, 662–675.
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890.
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533.
- Prestes, E.B.; Alves, L.S.; Rodrigues, D.A.S.; Dutra, F.F.; Fernandez, P.L.; Paiva, C.N.; Kagan, J.C.; Bozza, M.T. Mitochondrial Reactive Oxygen Species Participate in Signaling Triggered by Heme in Macrophages and upon Hemolysis. J. Immunol. 2020, 205, 2795–2805.
- Mittal, M.; Sánchez-Rodríguez, R.; Spera, I.; Venegas, F.C.; Favia, M.; Viola, A.; Castegna, A. Reactive Oxygen Species in Macrophages: Sources and Targets. Front. Immunol. 2021, 12, 734229.
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85.
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781.
- Zhao, J.; Pan, L.; Zhou, M.; Yang, Z.; Meng, Y.; Zhang, X. Comparative Physiological and Transcriptomic Analyses Reveal Mechanisms of Improved Osmotic Stress Tolerance in Annual Ryegrass by Exogenous Chitosan. Genes 2019, 10, 853.
- Fukai, T.; Ushio-Fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849.
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321.
- Mickiewicz, A.; Kreft, E.; Kuchta, A.; Wieczorek, E.; Marlęga, J.; Ćwiklińska, A.; Paprzycka, M.; Gruchała, M.; Fijałkowski, M.; Jankowski, M. The Impact of Lipoprotein Apheresis on Oxidative Stress Biomarkers and High-Density Lipoprotein Subfractions. Oxid. Med. Cell. Longev. 2020, 2020, 9709542.
- Gokce, N.; Keaney, J.F., Jr.; Hunter, L.M.; Watkins, M.T.; Nedeljkovic, Z.S.; Menzoian, J.O.; Vita, J.A. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events in patients with peripheral vascular disease. J. Am. Coll. Cardiol. 2003, 41, 1769–1775.
- Chirkov, Y.Y.; Nguyen, T.H.; Horowitz, J.D. Impairment of Anti-Aggregatory Responses to Nitric Oxide and Prostacyclin: Mechanisms and Clinical Implications in Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 1042.
- Shi, X.; Li, P.; Liu, H.; Prokosch, V. Oxidative Stress, Vascular Endothelium, and the Pathology of Neurodegeneration in Retina. Antioxidants 2022, 11, 543.
- Förstermann, U.; Li, H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br. J. Pharmacol. 2011, 164, 213–223.
- Kumar, S.; Verma, R.; Tyagi, N.; Gangenahalli, G.; Verma, Y.K. Therapeutics effect of mesenchymal stromal cells in reactive oxygen species-induced damages. Hum. Cell 2022, 35, 37–50.
- Nannelli, G.; Ziche, M.; Donnini, S.; Morbidelli, L. Endothelial Aldehyde Dehydrogenase 2 as a Target to Maintain Vascular Wellness and Function in Ageing. Biomedicines 2020, 8, 4.
- Nannelli, G.; Terzuoli, E.; Giorgio, V.; Donnini, S.; Lupetti, P.; Giachetti, A.; Bernardi, P.; Ziche, M. ALDH2 Activity Reduces Mitochondrial Oxygen Reserve Capacity in Endothelial Cells and Induces Senescence Properties. Oxid. Med. Cell Longev. 2018, 2018, 9765027.
- Wu, B.; Yu, L.; Wang, Y.; Wang, H.; Li, C.; Yin, Y.; Yang, J.; Wang, Z.; Zheng, Q.; Ma, H. Aldehyde dehydrogenase 2 activation in aged heart improves the autophagy by reducing the carbonyl modification on SIRT1. Oncotarget 2016, 7, 2175–2188.
- Infante, T.; Costa, D.; Napoli, C. Novel Insights Regarding Nitric Oxide and Cardiovascular Diseases. Angiology 2021, 72, 411–425.
- Farah, C.; Michel, L.Y.M.; Balligand, J.L. Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018, 15, 292–316.
- Rocha, B.S.; Gago, B.; Barbosa, R.M.; Cavaleiro, C.; Laranjinha, J. Ethyl nitrite is produced in the human stomach from dietary nitrate and ethanol, releasing nitric oxide at physiological pH: Potential impact on gastric motility. Free Radic. Biol. Med. 2015, 82, 160–166.
- Wan, S.H.; Pandey, A. Targeting the nitrate-nitrite-nitric oxide pathway in heart failure with preserved ejection fraction: Too soon to say no to nitric oxide? Eur. J. Heart Fail. 2021, 23, 824–825.
- Larsen, F.J.; Ekblom, B.; Sahlin, K.; Lundberg, J.O.; Weitzberg, E. Effects of dietary nitrate on blood pressure in healthy volunteers. N. Engl. J. Med. 2006, 355, 2792–2793.
- Weitzberg, E.; Lundberg, J.O. Novel aspects of dietary nitrate and human health. Annu. Rev. Nutr. 2013, 33, 129–159.
- Weitzberg, E.; Hezel, M.; Lundberg, J.O. Nitrate-nitrite-nitric oxide pathway implications for anesthesiology and intensive care. Anesthesiology 2010, 113, 1460–1475.
- Lundberg, J.O.; Weitzberg, E. NO-synthase independent NO generation in mammals. Biochem. Biophys. Res. Commun. 2010, 396, 39–45.
- Stokes, K.Y.; Dugas, T.R.; Tang, Y.; Garg, H.; Guidry, E.; Bryan, N.S. Dietary nitrite prevents hypercholesterolemic microvascular inflammation and reverses endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1281–H1288.
- Sindler, A.L.; Fleenor, B.S.; Calvert, J.W.; Marshall, K.D.; Zigler, M.L.; Lefer, D.J.; Seals, D.R. Nitrite supplementation reverses vascular endothelial dysfunction and large elastic artery stiffness with aging. Aging Cell 2011, 10, 429–437.
- Webb, A.J.; Patel, N.; Loukogeorgakis, S. Acute blood pressure lowering, vasoprotective, and antiplatelet properties of dietary nitrate via bioconversion to nitrite. Hypertension 2008, 51, 784–790.
- Wagner, D.A.; Schultz, D.S.; Carlstrom, M.; Persson, A.E.; Larsson, E. Dietary nitrate attenuates oxidative stress, prevents cardiac and renal injuries, and reduces blood pressure in salt-induced hypertension. Cardiovasc. Res. 2011, 89, 574–585.
- Gao, X.; Yang, T.; Liu, M. NADPH oxidase in the renal microvasculature is a primary target for blood pressure lowering effects by inorganic nitrate and nitrite. Hypertension 2015, 65, 161–170.
- Fleenor, B.S.; Seals, D.R.; Zigler, M.L.; Sindler, A.L. Superoxide-lowering therapy with TEMPOL reverses arterial dysfunction with aging in mice. Aging Cell 2012, 11, 269–276.
- Webb, A.; Bond, R.; McLean, P.; Uppal, R.; Benjamin, N.; Ahluwalia, A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc. Natl. Acad. Sci. USA 2004, 101, 13683–13688.
- Jung, K.H.; Chu, K.; Ko, S.Y. Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury. Stroke 2006, 37, 2744–2750.
- Tripatara, P.; Patel, N.S.; Webb, A. Nitrite-derived nitric oxide protects the rat kidney against ischemia/reperfusion injury in vivo: Role for xanthine oxidoreductase. J. Am. Soc. Nephrol. 2007, 18, 570–580.
- Duranski, M.R.; Greer, J.J.; Dejam, A. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J. Clin. Investig. 2005, 115, 1232–1240.
- Kumar, D.; Branch, B.G.; Pattillo, C.B. Chronic sodium nitrite therapy augments ischemia-induced angiogenesis and arteriogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 7540–7545.
- Shiva, S.; Sack, M.N.; Greer, J.J. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J. Exp. Med. 2007, 204, 2089–2102.
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759.
- Jones, D.A.; Pellaton, C.; Velmurugan, S. Randomized phase 2 trial of intra-coronary nitrite during acute myocardial infarction. Circ. Res. 2015, 116, 437–447.
- Jabs, A.; Oelze, M.; Mikhed, Y.; Stamm, P.; Kröller-Schön, S.; Welschof, P.; Jansen, T.; Hausding, M.; Kopp, M.; Steven, S.; et al. Effect of soluble guanylyl cyclase activator and stimulator therapy on nitroglycerin-induced nitrate tolerance in rats. Vascul. Pharmacol. 2015, 71, 181–191.
- Münzel, T.; Daiber, A. Inorganic nitrite and nitrate in cardiovascular therapy: A better alternative to organic nitrates as nitric oxide donors? Vascul. Pharmacol. 2018, 102, 1–10.
- Munzel, T.; Gori, T. Nitrate therapy and nitrate tolerance in patients with coronary artery disease. Curr. Opin. Pharmacol. 2013, 13, 251–259.
- Gao, J.; Hao, Y.; Piao, X.; Gu, X. Aldehyde Dehydrogenase 2 as a Therapeutic Target in Oxidative Stress-Related Diseases: Post-Translational Modifications Deserve More Attention. Int. J. Mol. Sci. 2022, 23, 2682.
- Marini, E.; Giorgis, M.; Rolando, B.; Chegaev, K.; Lazzarato, L.; Bertinaria, M.; Vincenti, M.; Di Stilo, A. Multitarget Antioxidant NO-Donor Organic Nitrates: A Novel Approach to Overcome Nitrates Tolerance, an Ex Vivo Study. Antioxidants 2022, 11, 166.
- Yager, N.; Konduru, S.; Torosoff, M. Nitrates as a Marker of Multiple Co-morbidities and Increased Mortality in Patients Undergoing Percutaneous Coronary Intervention (PCI). Cureus 2022, 14, e23520.
- Brandt, M.; Garlapati, V.; Oelze, M.; Sotiriou, E.; Knorr, M.; Kröller-Schön, S.; Kossmann, S.; Schönfelder, T.; Morawietz, H.; Schulz, E.; et al. NOX2 amplifies acetaldehyde-mediated cardiomyocyte mitochondrial dysfunction in alcoholic cardiomyopathy. Sci. Rep. 2016, 6, 32554.
- Roy, B.; Palaniyandi, S.S. Aldehyde dehydrogenase 2 inhibition potentiates 4-hydroxy-2-nonenal induced decrease in angiogenesis of coronary endothelial cells. Cell Biochem. Funct. 2020, 38, 290–299.
- Kang, P.; Wang, J.; Fang, D.; Fang, T.; Yu, Y.; Zhang, W.; Shen, L.; Li, Z.; Wang, H.; Ye, H.; et al. Activation of ALDH2 attenuates high glucose induced rat cardiomyocyte fibrosis and necroptosis. Free Radic. Biol. Med. 2020, 146, 198–210.
- Jang, A.J.; Lee, J.H.; Yotsu-Yamashita, M.; Park, J.; Kye, S.; Benza, R.L.; Passineau, M.J.; Jeon, Y.J.; Nyunoya, T. A Novel Compound, "FA-1" Isolated from Prunus mume, Protects Human Bronchial Epithelial Cells and Keratinocytes from Cigarette Smoke Extract-Induced Damage. Sci. Rep. 2018, 8, 11504.
- Yang, Y.; Chen, W.; Wang, X.; Ge, W. Impact of mitochondrial aldehyde dehydrogenase 2 on cognitive impairment in the AD model mouse. Acta Biochim. Biophys. Sin. 2021, 53, 837–847.
- Chu, A.; Najafzadeh, P.; Sullivan, P.; Cone, B.; Elshimali, R.; Shakeri, H.; Janzen, C.; Mah, V.; Wadehra, M. Aldehyde dehydrogenase isoforms and inflammatory cell populations are differentially expressed in term human placentas affected by intrauterine growth restriction. Placenta 2019, 81, 9–17.
- Huddle, B.C.; Grimley, E.; Buchman, C.D.; Chtcherbinine, M.; Debnath, B.; Mehta, P.; Yang, K.; Morgan, C.A.; Li, S.; Felton, J.; et al. Structure-Based Optimization of a Novel Class of Aldehyde Dehydrogenase 1A (ALDH1A) Subfamily-Selective Inhibitors as Potential Adjuncts to Ovarian Cancer Chemotherapy. J. Med. Chem. 2018, 61, 8754–8773.
- Puttini, S.; Plaisance, I.; Barile, L.; Cervio, E.; Milano, G.; Marcato, P.; Pedrazzini, T.; Vassalli, G. ALDH1A3 Is the Key Isoform That Contributes to Aldehyde Dehydrogenase Activity and Affects in Vitro Proliferation in Cardiac Atrial Appendage Progenitor Cells. Front. Cardiovasc. Med. 2018, 5, 90.
- Deza-Ponzio, R.; Herrera, M.L.; Bellini, M.J.; Virgolini, M.B.; Hereñú, C.B. Aldehyde dehydrogenase 2 in the spotlight: The link between mitochondria and neurodegeneration. Neurotoxicology 2018, 68, 19–24.
- Ding, J.; Yang, Z.; Ma, H.; Zhang, H. Mitochondrial Aldehyde Dehydrogenase in Myocardial Ischemic and Ischemia-Reperfusion Injury. Adv. Exp. Med. Biol. 2019, 1193, 107–120.
- Liu, X.Z.; Sun, X.; Shen, K.P.; Jin, W.J.; Fu, Z.Y.; Tao, H.R.; Xu, Z.X. Aldehyde dehydrogenase 2 overexpression inhibits neuronal apoptosis after spinal cord ischemia/reperfusion injury. Neural Regen. Res. 2017, 12, 1166–1171.
- Ferreira, J.C.; Mochly-Rosen, D. Nitroglycerin use in myocardial infarction patients. Circ. J. 2012, 76, 15–21.
- Lang, B.S.; Gorren, A.C.; Oberdorfer, G.; Wenzl, M.V.; Furdui, C.M.; Poole, L.B.; Mayer, B.; Gruber, K. Vascular bioactivation of nitroglycerin by aldehyde dehydrogenase-2: Reaction intermediates revealed by crystallography and mass spectrometry. J. Biol. Chem. 2012, 287, 38124–38134.
- Chen, Y.R.; Nie, S.D.; Shan, W.; Jiang, D.J.; Shi, R.Z.; Zhou, Z.; Guo, R.; Zhang, Z.; Li, Y.J. Decrease in endogenous CGRP release in nitroglycerin tolerance: Role of ALDH-2. Eur. J. Pharmacol. 2007, 571, 44–50.
- Daiber, A.; Wenzel, P.; Oelze, M.; Schuhmacher, S.; Jansen, T.; Münzel, T. Mitochondrial aldehyde dehydrogenase (ALDH-2)-maker of and marker for nitrate tolerance in response to nitroglycerin treatment. Chem. Biol. Interact. 2009, 178, 40–47.
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23.
- Manni, M.E.; Rigacci, S.; Borchi, E.; Bargelli, V.; Miceli, C.; Giordano, C.; Raimondi, L.; Nediani, C. Monoamine Oxidase Is Overactivated in Left and Right Ventricles from Ischemic Hearts: An Intriguing Therapeutic Target. Oxid. Med. Cell. Longev. 2016, 2016, 4375418.
- Xu, Y.; Yuan, Q.; Cao, S.; Cui, S.; Xue, L.; Song, X.; Li, Z.; Xu, R.; Yuan, Q.; Li, R. Aldehyde dehydrogenase 2 inhibited oxidized LDL-induced NLRP3 inflammasome priming and activation via attenuating oxidative stress. Biochem. Biophys. Res. Commun. 2020, 529, 998–1004.
- Ge, W.; Yuan, M.; Ceylan, A.F.; Wang, X.; Ren, J. Mitochondrial aldehyde dehydrogenase protects against doxorubicin cardiotoxicity through a transient receptor potential channel vanilloid 1-mediated mechanism. Biochim. Biophys. Acta 2016, 1862, 622–634.
- Pan, G.; Munukutla, S.; Kar, A.; Gardinier, J.; Thandavarayan, R.A.; Palaniyandi, S.S. Type-2 diabetic aldehyde dehydrogenase 2 mutant mice (ALDH 2*2) exhibiting heart failure with preserved ejection fraction phenotype can be determined by exercise stress echocardiography. PLoS ONE 2018, 13, e0195796.
- Roy, B.; Sundar, K.; Palaniyandi, S.S. 4-hydroxy-2-nonenal decreases coronary endothelial cell migration: Potentiation by aldehyde dehydrogenase 2 inhibition. Vascul. Pharmacol. 2020, 131, 106762.
- Hellenthal, K.E.M.; Brabenec, L.; Gross, E.R.; Wagner, N.M. TRP Channels as Sensors of Aldehyde and Oxidative Stress. Biomolecules 2021, 11, 1401.
- Papatheodorou, I.; Galatou, E.; Panagiotidis, G.D.; Ravingerová, T.; Lazou, A. Cardioprotective Effects of PPARβ/δ Activation against Ischemia/Reperfusion Injury in Rat Heart Are Associated with ALDH2 Upregulation, Amelioration of Oxidative Stress and Preservation of Mitochondrial Energy Production. Int. J. Mol. Sci. 2021, 22, 6399.
- Chen, C.H.; Ferreira, J.C.B.; Mochly-Rosen, D. ALDH2 and Cardiovascular Disease. Adv. Exp. Med. Biol. 2019, 1193, 53–67.
- Chang, B.; Hao, S.; Zhang, L.; Gao, M.; Sun, Y.; Huang, A.; Teng, G.; Li, B.; Crabb, D.W.; Kusumanchi, P.; et al. Association Between Aldehyde Dehydrogenase 2 Glu504Lys Polymorphism and Alcoholic Liver Disease. Am. J. Med. Sci. 2018, 356, 10–14.
- Chen, C.C.; Lu, R.B.; Chen, Y.C.; Wang, M.F.; Chang, Y.C.; Li, T.K.; Yin, S.J. Interaction between the functional polymorphisms of the alcohol-metabolism genes in protection against alcoholism. Am. J. Hum. Genet. 1999, 65, 795–807.
- Jung, S.J.; Hwang, J.H.; Park, E.O.; Lee, S.O.; Chung, Y.J.; Chung, M.J.; Lim, S.; Lim, T.J.; Ha, Y.; Park, B.H.; et al. Regulation of Alcohol and Acetaldehyde Metabolism by a Mixture of Lactobacillus and Bifidobacterium Species in Human. Nutrients 2021, 13, 1875.
- He, J.D.; Lytvyn, Y.; Zhou, K.; Parker, J.D. Role of Mitochondrial Aldehyde Dehydrogenase in Nitroglycerin-Mediated Vasodilation: Observations Concerning the Dose-Response Relationship. J. Cardiovasc. Pharmacol. 2019, 73, 359–364.
- Yamaki, N.; Matsushita, S.; Hara, S.; Yokoyama, A.; Hishimoto, A.; Higuchi, S. Telomere shortening in alcohol dependence: Roles of alcohol and acetaldehyde. J. Psychiatr. Res. 2019, 109, 27–32.
- Miura, T.; Nishinaka, T.; Terada, T.; Yonezawa, K. Vasodilatory effect of nitroglycerin in Japanese subjects with different aldehyde dehydrogenase 2 (ALDH2) genotypes. Chem. Biol. Interact. 2017, 276, 40–45.
- Mizuno, Y.; Harada, E.; Kugimiya, F.; Shono, M.; Kusumegi, I.; Yoshimura, M.; Kinoshita, K.; Yasue, H. East Asians Variant Mitochondrial Aldehyde Dehydrogenase 2 Genotype Exacerbates Nitrate Tolerance in Patients With Coronary Spastic Angina. Circ. J. 2020, 84, 479–486.
- Zhao, Y.; Wang, C. Glu504Lys Single Nucleotide Polymorphism of Aldehyde Dehydrogenase 2 Gene and the Risk of Human Diseases. Biomed. Res. Int. 2015, 2015, 174050.
- Pan, G.; Deshpande, M.; Pang, H.; Palaniyandi, S.S. Precision medicine approach: Empagliflozin for diabetic cardiomyopathy in mice with aldehyde dehydrogenase (ALDH) 2*2 mutation, a specific genetic mutation in millions of East Asians. Eur. J. Pharmacol. 2018, 839, 76–81.
- Hu, Y.F.; Wu, C.H.; Lai, T.C.; Chang, Y.C.; Hwang, M.J.; Chang, T.Y.; Weng, C.H.; Chang, P.M.; Chen, C.H.; Mochly-Rosen, D.; et al. ALDH2 deficiency induces atrial fibrillation through dysregulated cardiac sodium channel and mitochondrial bioenergetics: A multi-omics analysis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166088.
- Leo, C.H.; Fernando, D.T.; Tran, L.; Ng, H.H.; Marshall, S.A.; Parry, L.J. Serelaxin Treatment Reduces Oxidative Stress and Increases Aldehyde Dehydrogenase-2 to Attenuate Nitrate Tolerance. Front. Pharmacol. 2017, 8, 141.
More