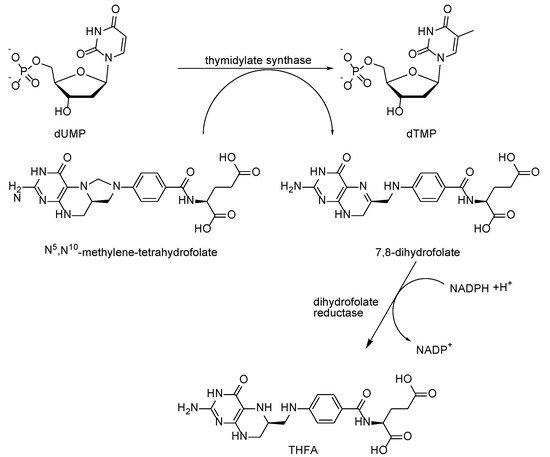
2. Mechanism of Antifolates Action
Folic acid (
1) first has to be reduced to THFA (
2) by dihydrofolate reductase, after which it can attach various one-carbon groups and transfer them to other molecules. In the reaction, once catalyzed by thymidylate synthase, deoxyuridine monophosphate (deoxy-UMP or dUMP) is converted to deoxythymidine monophosphate (deoxy-TMP or dTMP), producing a methylene group from 5,10-methylene-THFA; the latter is oxidized into dihydrofolic acid and must be reduced again to participate in further reactions. Methotrexate (MTX) and other folic acid antagonists with a high affinity for dihydrofolate reductase (K, 0.01–0.2 nmol/L) disrupt the formation of THFA, causing a deficiency of reduced folates and an accumulation of toxic dihydrofolic acid polyglutamates. At the same time, the transfer reactions of one-carbon groups, which are necessary for the synthesis of purines and dTMP, are inhibited; as a result, the synthesis of nucleic acids and other metabolic processes are disrupted. The toxic action of methotrexate is prevented by calcium folinate (the calcium salt of 5-formyl-THFA), which enters the cell via a reduced folate transporter and is converted into the other THFA derivatives [
14] (
Scheme 1).
Scheme 1. Main target processes for antifolates in living cells.
Once it became clear that methotrexate directly inhibits not only dihydrofolate reductase but also the enzymes for the synthesis of purines and thymidylate synthase, the coenzymes of which are reduced folates, a search commenced for folic acid antagonists that selectively inhibit these enzymes. By replacing the N-5, N-8 and N-10 atoms and modifying the side chains of the methotrexate molecule, it was possible to synthesize drugs that retain their inherent ability to form stable polyglutamates inside the cell, but better penetrate the tumor [
15], such as the following: raltitrexed, a thymidylate synthase inhibitor; lometrexol, a purine synthesis inhibitor; and pemetrexed, which combines both mechanisms of action [
16].
Most folic acid antimetabolites are only partially selective for tumor cells and affect rapidly proliferating normal cells, including bone marrow and gastrointestinal mucosa. Folic acid antagonists act in the S-period and are most active against cells in the logarithmic growth phase [
17].
3. Methotrexate: (S)-2-(4-(((2,4-Diaminopteridin-6-yl)Methyl)(Methyl)Amino)benzamido) Pentanedioic Acid (MTX, Rheumatrex, Amethopterin, Abitrexate, Trexall, Methylaminopterin, Mexate, Metatrexan)
The discovery of the first folic acid antagonist, methotrexate (MTX), with its promising activity for the treatment of a variety of human cancers, prompted the search for other folate analogs [
18]. As a structural analogue of folic acid, methotrexate inhibits the activity of the enzyme folate reductase, which prevents the conversion of folic acid into tetrahydrofolic acid, which is involved in cell metabolism and reproduction. Methotrexate is recommended for acute childhood leukemia; chorionepithelioma of the uterus; cancer of the breast, lungs, testicles, and other malignant tumors in adults (in combination with other antiblastoma drugs); and is also used as an immunosuppressive agent.
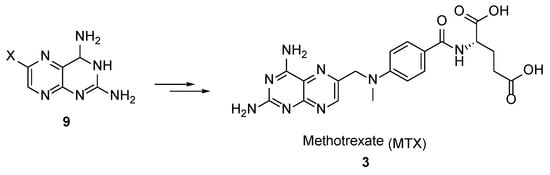
The most common synthetic strategy for the preparation of MTX
3 involves the post-modification of 3,4-dihydropteridine-2,4-diamines
9, as depicted in
Scheme 2.
Scheme 2. Synthetic strategy towards MTX by the post-modification of 3,4-dihydropteridine-2,4-diamines 9.
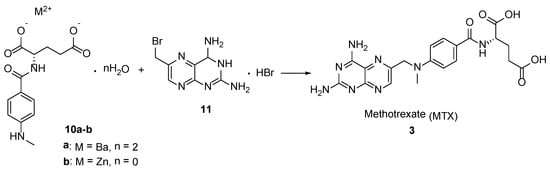
Thus, MTX was obtained by the reaction of 2,4-diamino-6-bromomethylpteridine hydrobromide
11 with barium salt dehydrate [
19] or Zn
2+ salt [
20] of
p-(
N-methyl)-aminobenzoyl-
L-glutamic acid
10 in 87.5% and 56.1% yields, accordingly (
Scheme 3).
Scheme 3. Synthesis of MTX by the reaction between metal salt of p-(N-methyl)-aminobenzoyl-L-glutamic acid and 2,4-diamino-6-bromomethylpteridine hydrobromide.
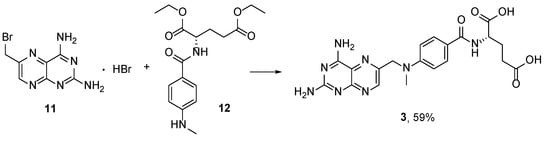
The reaction of
11 with the diethyl
p-(
N-methyl)-aminobenzoyl-
L-glutamate
12 followed by basic saponification (
Scheme 4) provided lower yields of the target product [
21].
Scheme 4. Synthesis of MTX by the reaction of 2,4-diamino-6-bromomethylpteridine hydrobromide 11 with diethyl p-(N-methyl)-aminobenzoyl-L-glutamate 12.
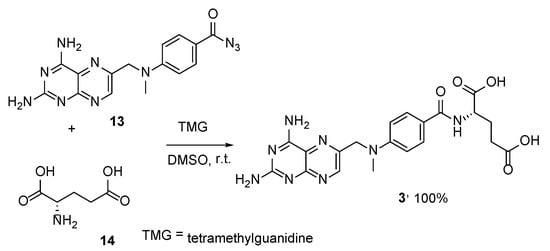
Another approach involves the substitution of the azide group in 4-(
N-methyl-
N′-(6″-aminopteroil-methyleno)aminobenzoic acid derivative
13 in a reaction with L-glutamic acid
14 in DMSO at room temperature in the presence of tetramethylguanidine (TMG) as the base (
Scheme 5) [
22]. The reaction resulted in the corresponding desired MTX in a quantitative yield, which is the main advantage of this method.
Scheme 5. Quantitative synthesis of MTX by the reaction of 4-(N-methyl-N′-(6″-aminopteroil-methyleno)aminobenzoic acid derivative 13 with L-glutamic acid 14.
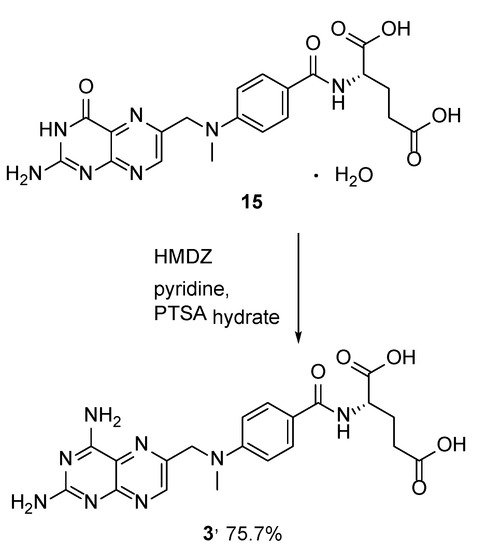
In addition, MTX was obtained in a 75.7% yield by means of the transformation of its more stable and synthetically available 4-oxoderivative (methopterin hydrate) (
15) in the presence of pyridine,
p-toluenesulfonic acid monohydrate and 1,1,1,3,3,3-hexamethyl-disilazane (HMDZ) (
Scheme 6) [
23].
Scheme 6. Synthesis of MTX from 4-oxoderivative of MTX (methopterin hydrate) (15).
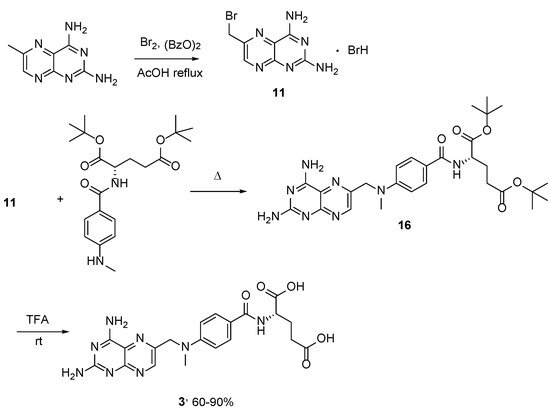
Along with MTX, its
13C-multilabelled forms with
13C-enrichment at 2, 7, 9, 4, 7, 8a, 9 and 2, 4a,b positions were synthesized from the di-
tert-butyl ester of MTX
16 for the NMR study of the mechanisms of drug–enzyme interactions (
Scheme 7) [
24]. The reaction was carried out by performing ‘benzylic’ bromination, followed by the substitution of the bromine atom by the di-
t-butyl
N-(
p-methylaminobenzoyl)-
L-glutamate. The acid treatment of each of the formed methotrexate di-
t-butyl esters yielded the corresponding
13C-enriched methotrexate in 60–90% yields. So far, this is the only method reported for the synthesis of C
13-MTX.
Scheme 7. Multistep synthesis of MTX.
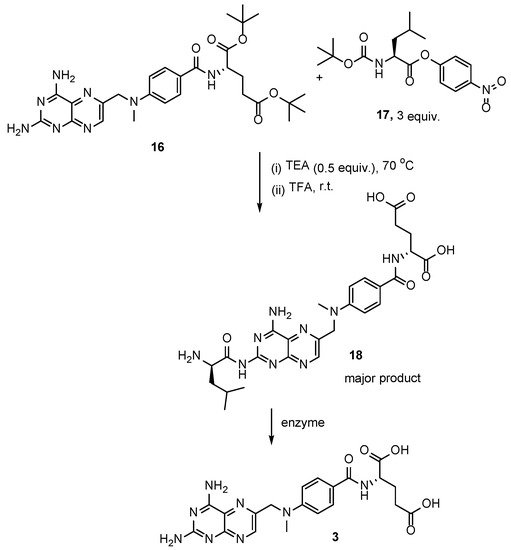
In another method for the synthesis of MTX
3, the pro-drug of MTX,
N-(
L-α-aminoacyl)-derivative of methotrexate
18, was initially prepared by a reaction between the di-
tert-butyl ester of MTX
16 and
N-
tert-butyloxycarbonyl-
L-leucine derivative
17, followed by the acidic deprotection of protective groups [
25]. Subsequently, the obtained pro-drug
18 was successfully converted into MTX via the enzymatic cleavage by porcine microsomal leucine aminopeptidase (
Scheme 8). Unfortunately, the authors did not provide any yields due to the format of the publication.
Scheme 8. Three-step synthesis of MTX starting from di-tert-butyl ester of MTX 16 and N-tert-butyloxycarbonyl-L-leucine derivative 17.
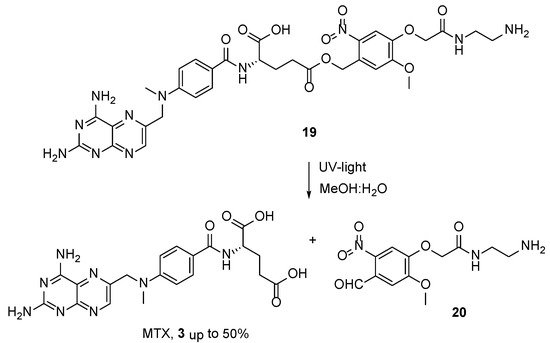
Free-form MTX was obtained from the conjugate of the
o-nitrobenzyl alcohol derivative and MTX
19 during a photolysis experiment in aqueous methanol under UV-light irradiation [
26]. This technique was considered by the authors as a possible way to transport the MTX to the cancer cells with the release of MTX free form at up to 50% at a pH level of 7.4 (
Scheme 9).
Scheme 9. UV-light promoted synthesis of MTX from
19. Reproduced with the permission of reference [
26]. Copyright © 2011, Elsevier Ltd.
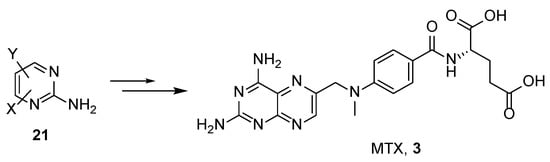
In the literature, there are less common synthetic approaches available that involve the construction of a 3,4-dihydropteridine core starting from aminopyrimidines
21 (
Scheme 10).
Scheme 10. Synthetic strategy towards MTX by the post-modification of aminopyrimidines 21.
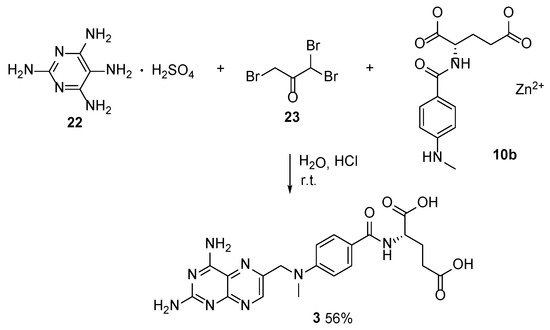
In this context, MTX was obtained by the tandem multicomponent reaction between Zn
2+ salt of
N-(4-
N-methylaminobenzoil)-
L-glutamic acid
10b, 1,1,3-tribromoacetone
23 and 2,2,5,6-tetraaminopyrimidine sulfate
22 under mild conditions (
Scheme 11) [
27]. This method has a noticeable advantage, such as the possibility to carry out several reactions in one step without the isolation of intermediates during each step.
Scheme 11. Multicomponent synthesis of MTX under mild reaction conditions.
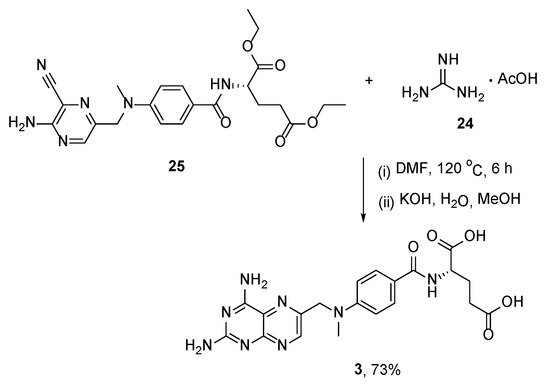
In another method, the MTX core was constructed by means of a heterocyclization reaction between commercially available guanidine acetate
24 and easily derived diethyl (4-(((5-amino-6-cyanopyrazin-2-yl)methyl)(methyl)amino)benzoyl)glutamate
25 under heating conditions, followed by basic hydrolysis (
Scheme 12) [
28].
Scheme 12. Synthesis of MTX by the reaction between guanidine acetate 24 and diethyl (4-(((5-amino-6-cyanopyrazin-2-yl)methyl)(methyl)amino)benzoyl)glutamate 25.
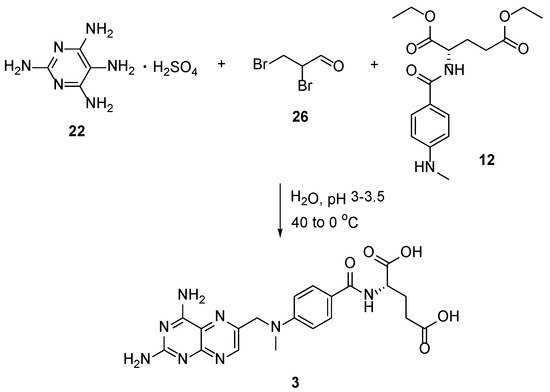
Lastly, the approach for MTX
3 involves a reaction between 2,4,5,6-tetraaminopyrimidine hydrosulphate
22, 2,3-dibromopropionaldehyde
26, and
N-4-(methylamino)benzoyl)-
L-glutamic acid
12 disodium salt under oxidative conditions (iodine in the presence of KI) (
Scheme 13) [
29]. In this article, the authors were more concerned about the purity of the obtained compounds than their yields.
Scheme 13. Synthesis of MTX by the reaction between 2,4,5,6-tetraaminopyrimidine hydrosulphate 22, 2,3-dibromopropionaldehyde 26, and N-4-(methylamino)benzoyl)-L-glutamic acid 12 disodium salt under oxidative conditions.
4. Raltitrexed: (2S)-2-[[5-[Methyl-[(2-Methyl-4-oxo-3H-Quinazolin-6-yl)Methyl]Amino] Thiophene-2-Carbonyl]Amino]Pentanedioic Acid (Tomudex, ZD1694)
Raltitrexed (Tomudex) is a more recent, specific, mixed, and non-competitive inhibitor of thymidylate synthase indicated for use in cancer therapy, especially colorectal cancer [
30,
31,
32].
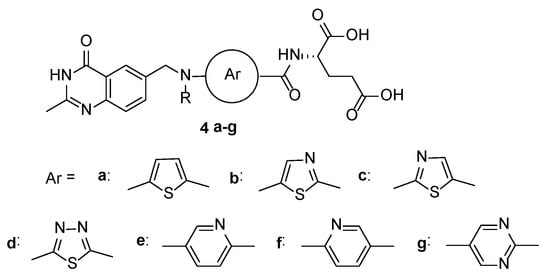
In 1991, Marsham et al. reported the synthesis of a series of C2-methyl-N10-alkylquinazoline-based antifolates, in which the benzene ring was replaced by the heterocycles, i.e., thiophene, thiazole, thiadiazole, pyridine, and pyrimidine (
Scheme 14) [
33].
Scheme 14. Structures of a series of C2-methyl-N10-alkylquinazoline-based antifolates. Reproduced with the permission of reference [
33]. Copyright © 1991, American Chemical Society.
The thiophene system
4a and its related thiazole
4b yielded analogues that were considerably more efficient than the parent benzene series as inhibitors of L1210 cell growth. Although, in general, these heterocycles were somewhat poorer inhibitors of the isolated TS enzyme. Raltitrexed
4a (R = CH
3) was synthesized in a 41% yield starting with the thiophene-2-carboxylic acid, as shown in
Scheme 15.
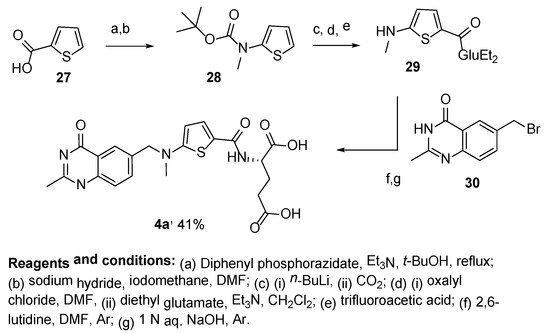
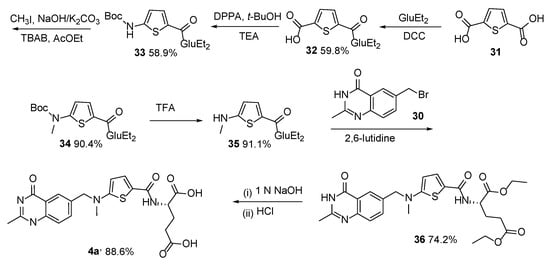
Scheme 15. Multistep synthesis of Raltitrexed 4a starting from thiophene-2-carboxylic acid 27.
Another route to Raltitrexed was reported that started with thiophene-2,5-dicarboxylic acid
31, which was then converted in four steps to diethyl (5-(methylamino)thiophene-2-carbonyl)-
L-glutamate
35. This was followed by an alkylation reaction of the last one with 6-bromomethyl-2-methyl-4-quinazolinone
30 and basic hydrolysis, which resulted in the target product
4a (
Scheme 16) [
34].
Scheme 16. Multistep synthesis of Raltitrexed starting from thiophene-2,5-dicarboxylic acid 31.
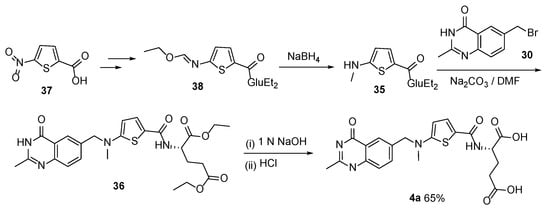
A similar route to Raltitrexed was reported by Yao et al. starting with 5-nitrothiophene-2-carboxylic acid
37 via the sequence of NaBH
4 reduction, alkylation, and saponification (
Scheme 17) [
35]. The target product was isolated in a lower yield.
Scheme 17. Multistep synthesis of Raltitrexed starting from 5-nitrothiophene-2-carboxylic acid 37.
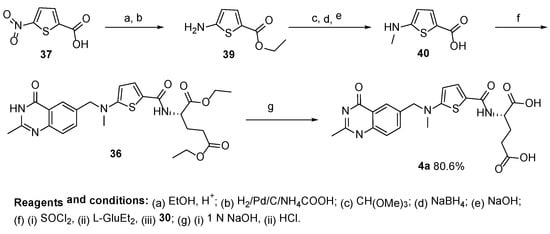
Raltitrexed was also prepared by using the same compound in less reaction steps as reported by Xiqun et al. (
Scheme 18) [
36].
Scheme 18. Multistep synthesis of Raltitrexed starting from 5-nitrothiophene-2-carboxylic acid 37.
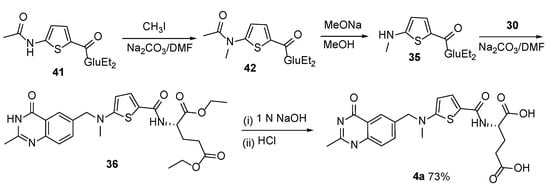
Moreover, the most recent and—in our opinion—easiest approach was reported in the work of H. Shaojie et al. regarding Raltitrexed, which involves a four-step sequence using diethyl (5-(
N-methylacetamido)thiophene-2-carbonyl)-
L-glutamate
41 as the starting material (
Scheme 19) [
37].
Scheme 19. Synthesis of Raltitrexed by diethyl (5-(N-methylacetamido)thiophene-2-carbonyl)-L-glutamate 41 as the starting material.
5. Pralatrexate (Folotyn): N-4-[1-(2,4-Diaminopteridin-6-yl)Pent-4-yn-2-yl]Benzoyl-L-Glutamic Acid
Pralatrexate
25 is another folate antagonist and antineoplastic agent with confirmed activity for the treatment of relapsed or refractory peripheral T-cell lymphoma (PTCL). Pralatrexate was approved for medical use in the United States in September 2009, as the first treatment for Peripheral T-cell Lymphoma (PTCL) [
38,
39], an often-aggressive type of non-Hodgkins lymphoma [
40].
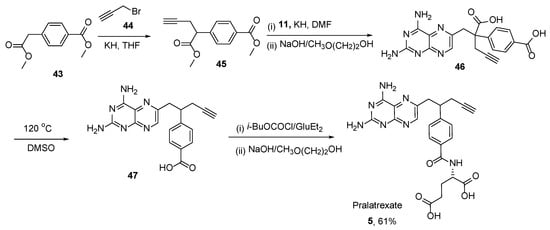
Successive alkylation of dimethyl homoterephthalate
43 with propargyl bromide
44 and 2,4-diamino-6-(bromomethy1)pteridine
11 followed by ester saponification at room temperature resulted in 2,4-diamino-4-deoxy-10-carboxy-10-propargy1-10-deazapteroic acid
46. Subsequently, compound
46 was readily decarboxylated by heating in DMSO at 120 °C to yield diamino-10-propargyl-10-deazapteroic acid
47 as a precursor of Pralatrexate
5. Additionally, the coupling of
47 with diethyl
L-glutamate followed by ester hydrolysis, yielded Pralatrexate
5 (
Scheme 20) [
41].
Scheme 20. Synthesis of Pralatrexate starting from the reaction between dimethyl homoterephthalate
43 and propargyl bromide. Reproduced with the permission of reference [
41]. Copyright © 1993, American Chemical Society.
After the abovementioned publication, many improved procedures were reported for the preparation of Pralatrexate [
42,
43,
44,
45,
46,
47,
48]. The synthesis of optically pure diastereomers of Pralatrexate has also been reported [
49].
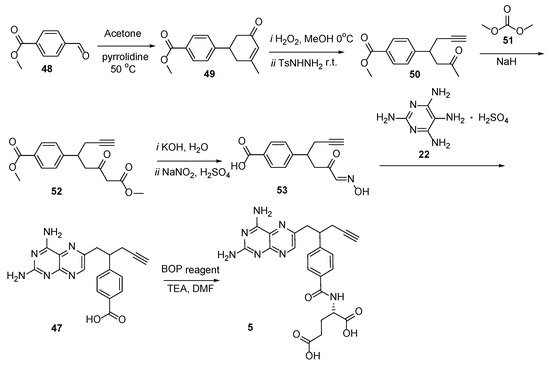
Another approach to producing Pralatrexate was developed by Alla et al. (2013), starting with ethyl 4-formylbenzoate
48; however, the yield was not specified (
Scheme 21) [
50].
Scheme 21. Multistep synthesis of Pralatrexate starting from ethyl 4-formylbenzoate 48.
2.5. Pemetrexed: (S)-2-(4-(2-(2-Amino-4-oxo-4,7-Dihydro-1H-Pyrrolo[2,3-d]Pyrimidin-5-yl)Ethyl)Benzamido)Pentanedioic Acid (PMX, ALIMTA, LY231514, MTA)
Pemetrexed (PMX)
30 is a folate antagonist and antineoplastic agent, used in the treatment of non-small cell lung cancer [
51,
52,
53,
54] and malignant mesothelioma [
55]. The mechanism of action of PMX is based on the inhibition of three enzymes responsible for the purine and pyrimidine synthesis—thymidylate synthase (TS), dihydrofolate reductase (DHFR), and glycinamide ribonucleotide formyltransferase [
56]—which prevents the formation of DNA and RNA, which are responsible for the growth of normal and cancer cells.
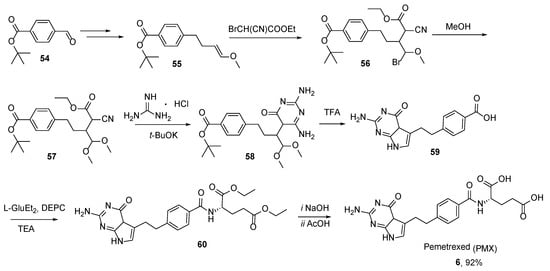
The first synthetic approach toward PMX was reported starting with
tert-butyl-4-formylbenzoate
54. This aliphatic precursor was heterocyclized to the PMX diethyl ester
60 in a few steps, which was converted to PMX
6 by performing hydrolysis (
Scheme 22) [
57].
Scheme 22. Multistep synthesis of Pemetrexed starting from tert-butyl-4-formylbenzoate 54.
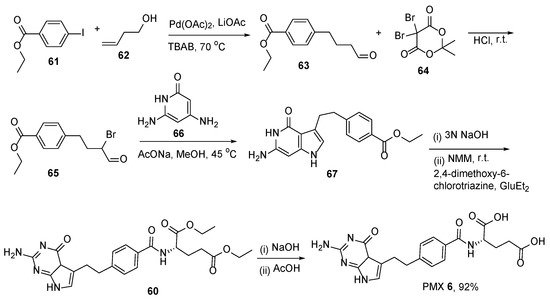
Mitchell-Ryan et al. reported the synthesis of 5-substituted pyrrolo[2,3-
d]pyrimidine antifolates with one-to-six bridge carbons and a benzoyl ring in the side chain as antitumor agents [
58]. The compound with a 4-carbon bridge was the most active analogue and it potentially inhibited the proliferation of the folate receptor (FR) α-expressing Chinese hamster ovary and KB human tumor cells. PMX was synthesized from ethyl 4-iodobenzoate
61, and 1-butene-4-ol
62 using a Heck cross-coupling reaction followed by bromination of the aldehyde at alpha-position. Further heterocyclization with basic hydrolysis and the formation of amide from diethyl-
L-glutamate resulted in acid derivative
60. In the final step, PMX
6 was obtained by the basic hydrolysis of the ester groups in a glutamate moiety (
Scheme 23).
Scheme 23. Synthesis of Pemetrexed starting from ethyl 4-iodobenzoate
61, and 1-butene-4-ol
62 through Heck cross-coupling reaction. Reproduced with the permission of reference [
58]. Copyright © 2013, American Chemical Society.
As an improvement to the abovementioned method, the preparation of lysin salt of PMX was reported [
59].
Michalak et al. reported the synthesis of PMX along with its common impurities/side products, starting with 4-[2-(2-amino-4-oxo-4,7-dihydro-1
H-pyrrolo[2,3-
d]pyrimidin-5-yl)ethyl]benzoic acid
28 [
60].
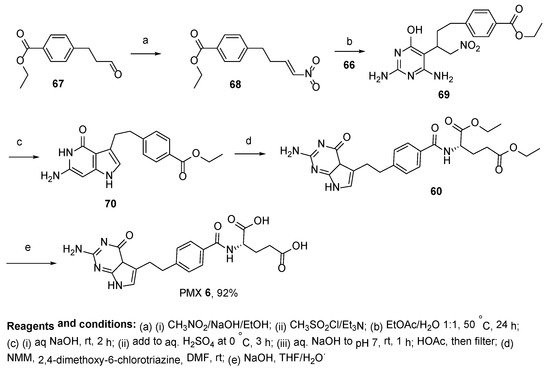
In the method reported by Tailor et al. for the synthesis of PMX, ethyl-4-(3-oxopropyl)benzoate
67 was used as a starting compound [
61,
62,
63]. After the Henry reaction with nitromethane, the product was converted to the semi-product with 2,6-diaminopyrimidin-4-ol
69. The heterocyclization of this semi-product resulted in pyrrolo[2,3-
d]pyrimidine derivative
70, which, followed by its functionalization with diethyl-
L-glutamate and basic hydrolysis, resulted in the desired product
6 in a 92% yield (
Scheme 24).
Scheme 24. Synthesis of Pemetrexed starting from ethyl-4-(3-oxopropyl)benzoate 67.
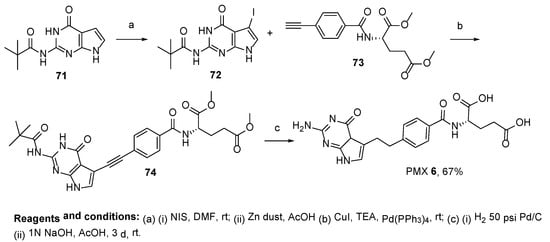
The same research group reported an improved synthesis of PMX, starting from dimethyl (4-ethynylbenzoyl)-
L-glutamate
73 and
N-(4-oxo-4,7-dihydro-3
H-pyrrolo[2,3-
d]pyrimidin-2-yl)pivalamide
71 [
64]. The sequence of iodination, reduction, Sonogashira cross-coupling, reduction reactions, and basic hydrolysis in the last step, resulted in the final product, PMX, in a 67% yield (
Scheme 25).
Scheme 25. Synthesis of Pemetrexed starting from dimethyl (4-ethynylbenzoyl)-L-glutamate 73 and N-(4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-2-yl)pivalamide 71.
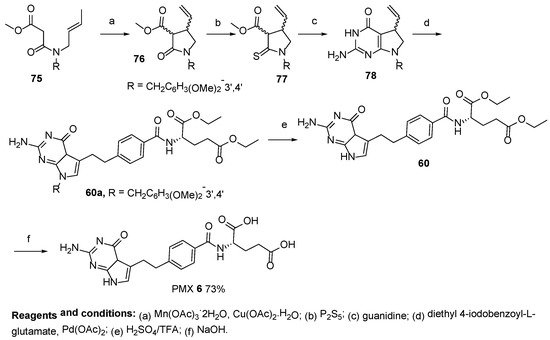
The same authors also reported the synthesis of PMX starting from methyl (
E)-3-(but-2-en-1-yl(3,4-dimethoxybenzyl)amino)-3-oxopropanoate [
65]. The last one was cyclized to methyl-1-(3,4-dimethoxybenzyl)-2-oxo-4-vinylpyrrolidine-3-carboxylate by the reaction of Mn(III) and Cu(II) acetates. The oxo-group was then converted to the thioxo-group upon treatment with P
2S
5. After the heterocyclization reaction, the obtained 2-amino-7-(3,4-dimethoxybenzyl)-5-vinyl-4
a,5,6,7-tetrahydro-4
H-pyrrolo[2,3-
d]pyrimidin-4-one was subjected to a Heck cross-coupling reaction with diethyl 4-iodobenzoylglutamate. Additionally, the coupling product was identified as one with unexpected double bond migration products in vinyl-bridged pyrrolinopyrimidine to form the ethano-bridged pyrrolopyrimidine. Thus, the authors avoided the reduction of the unsaturated bridge and the subsequent oxidation of the pyrroline ring at the same time. According to the authors, the protection of the N-7 position eliminates the PMX cell growth’s inhibitory activity. In addition, deprotection of the N-7 position was finally achieved upon treatment with a H
2SO
4/TFA mixture to facilitate the PMX precursor in a 30% yield, which resulted in the target product after saponification (
Scheme 26).
Scheme 26. Synthesis of Pemetrexed starting from methyl (E)-3-(but-2-en-1-yl(3,4-dimethoxybenzyl)amino)-3-oxopropanoate 75.
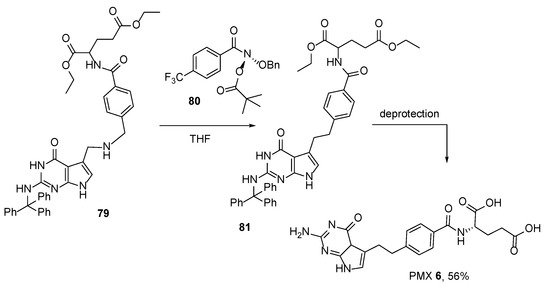
Finally, very recently, a method for PMX synthesis was developed by means of the reaction of an anomeric amide agent with a secondary amine precursor followed by the deprotection of protective groups (
Scheme 27) [
66].
Scheme 27. Synthesis of Pemetrexed by the reaction of anomeric amide agent with secondary amine precursor followed by the deprotection of protective groups.
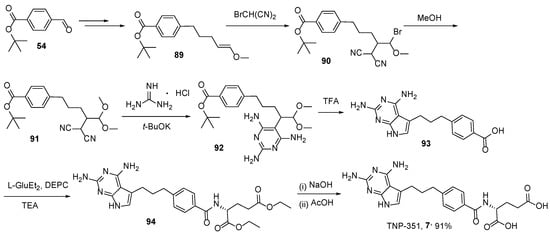
7. TNP-351: (2S)-2-[[4-[3-(2,4-Diamino-7H-Pyrrolo[2,3-d]Pyrimidin-5-yl)Propyl]benzoyl]Amino]Pentanedioic Acid (HY-19095)
TNP-351 is another antifolate from the same family as PMX. As a dihydrofolate reductase (DHFR) inhibitor, TNP-351 has good potential for the treatment of not only leukemia cells but also solid tumor cells, both in vitro and in vivo [
67]. The structure of TNP-351 contains three methylene bridges instead of two as in PMX and two amino groups in pyrimidine core.
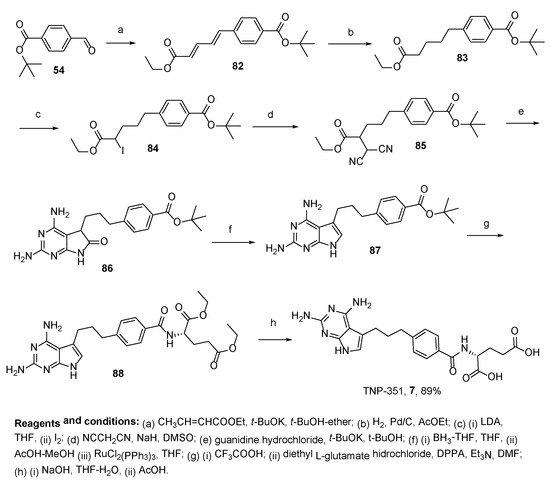
So far, only two synthetic approaches to TNP-351
7 have been reported; the first one includes construction of the key intermediary acyclic skeleton, 5-[4-(
tert-butoxycarbonyl)phenyl]-2-(dicyanomethyl)pentanoate
85, cyclization with guanidine, followed by reduction to pyrrolo[2,3-
d]pyrimidine derivatives
87, and subsequent glutamate coupling and saponification. These antifolates were more growth-inhibitory by approximately one order of magnitude than methotrexate (MTX) against KB human epidermoid carcinoma cells and A549 human non-small cell lung carcinoma cells with in vitro culture (
Scheme 28) [
68].
Scheme 28. Multistep synthesis of TNP-351.
The second method belongs to the same article, where the synthesis of TNP-351 has been reported along with PMX synthesis (
Scheme 29) [
57].
Scheme 29. Multistep synthesis of TNP-351.
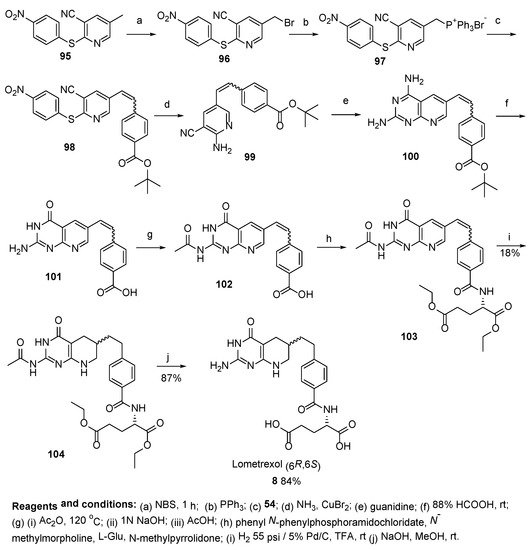
8. Lometrexol: (2S)-2-[[4-[2-[(6R)-2-Amino-4-oxo-5,6,7,8-Tetrahydro-1H-Pyrido[2,3-d]Pyrimidin-6-yl]Ethyl]Benzoyl]Amino]Pentanedioic Acid (LY 264618, DDATHF-B, Lometrexolum)
Lometrexol (6
R)-
8 is a folate analogue antimetabolite with antineoplastic activity [
69,
70,
71]. As the 6
R diastereomer of 5,10-dideazatetrahydrofolate, lometrexol inhibits glycinamide ribonucleotide formyltransferase (GARFT), the enzyme that catalyzes the first step in the de novo purine biosynthetic pathway, thereby inhibiting DNA synthesis, arresting cells in the S phase of the cell cycle, and inhibiting tumor cell proliferation. The agent is active against tumors that are resistant to the folate antagonist methotrexate.
Lometrexol has been used in trials for the treatment of lung cancer, drug/agent toxicity by tissues/organs, as well as for the treatment of unspecified adult solid tumors.
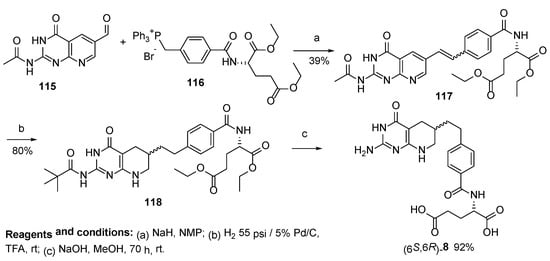
Taylor et al. reported several approaches to Lometrexol. The first of their approaches relates to the synthesis of (mixture of diastereomers)
(6S,6R)-Lometrexol
8 with a satisfactory yield starting from 5-methyl-2-((4-nitrophenyl)thio)nicotinonitrile
95 (
Scheme 30) [
72].
Scheme 30. Synthesis of Lometrexol starting from 5-methyl-2-((4-nitrophenyl)thio)nicotinonitrile 95.
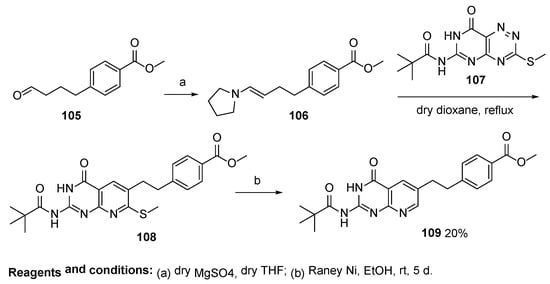
A key intermediate
109 for the subsequent synthesis of
(6S,6R)-Lometrexol was also prepared by Taylor et al. via a regiospecific intermolecular inverse electron demand Diels-Alder reaction between fused 1,2,4-triazines, 2-
N-pivaloyl-7-substituted-6-azapterins, and enamine (
Scheme 31) [
73].
Scheme 31. Synthesis of key intermediate 109.
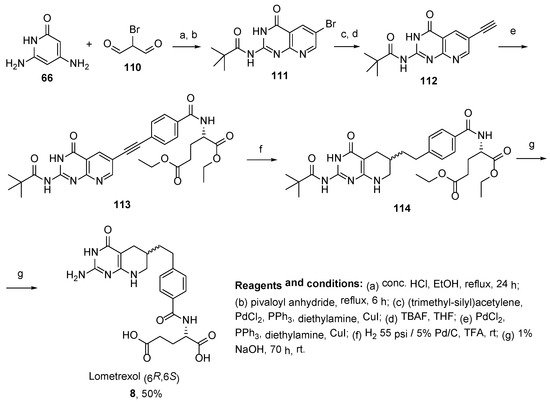
In another work, Taylor et al. developed a convenient method for the synthesis of
(6S,6R)-Lometrexol
8 with good yield via
N-(6-bromo-4-oxo-3,4-dihydropyrido[2,3-
d]pyrimidin-2-yl)pivalamide starting with 2,6-diaminopyrimidin-4(3
H)-one (
Scheme 32) [
74].
Scheme 32. Synthesis of (6S,6R)-Lometrexol 8 via N-(6-bromo-4-oxo-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)pivalamide starting from 2,6-diaminopyrimidin-4(3H)-one.
Boschelli et al. performed Wittig olefination of 2-acetyl-6-formyl-5-deazapterine to prepare
(6S,6R)-Lometrexol in three synthetic steps instead of Sonogashira cross-coupling of 2-pyvaloyl-6-formyl-5-deazapterine (
Scheme 33) [
75].
Scheme 33. Synthesis of (6S,6R)-Lometrexol 8 by the Wittig olefination of 2-acetyl-6-formyl-5-deazapterine.
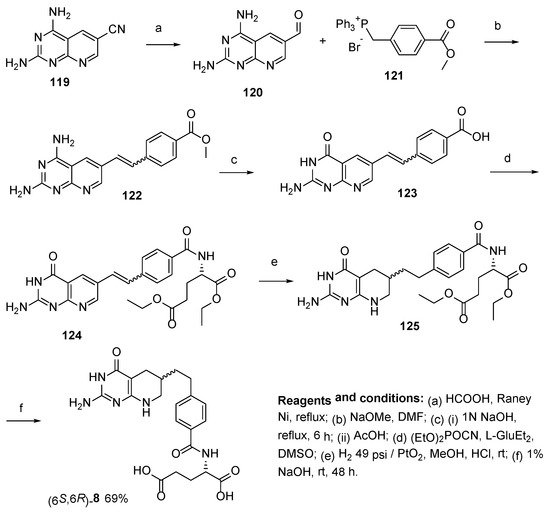
Similarly, Wittig olefination was used by Piper et al. for the synthesis of (6
S,6
R)-Lometrexol starting from 2,4-diaminopyrido[2,3-
d]pyrimidine-6-carboxaldehyde
120, derived from 6-carbonytrile, and [4-(methoxycarbonyl)benzylidene]triphenylphosphorane to yield 9,10-ethenyl precursor
122 [
76]. Standard hydrolytic deamination produced 5,10-dideazafolic acid
123, which was further converted to 5,10-dideazaaminopterin via a coupling reaction with dimethyl
L-glutamate by using (EtO)
2POCN, followed by hydrogenation and ester hydrolysis which led to the final product
8 (
Scheme 34).
Scheme 34. Synthesis of (6S,6R)-Lometrexol starting from 2,4-diaminopyrido[2,3-d]pyrimidine-6-carboxaldehyde.
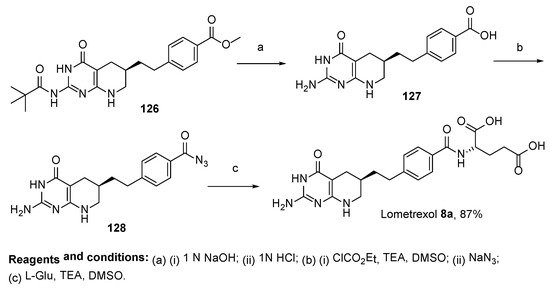
Currently, only two synthetic approaches toward diastereometrically pure 6
R-Lometrexol are reported in the literature. In this context, the synthesis of 6
R-Lometrexol was carried out starting from a double deprotected DDAH
4Pte–OH
126, which was obtained with preparative chiral-HPLC in a mixture of diastereomers derived from the route based on the work of Taylor et al. [
77]. After the transformation of the benzoic acid residue to a derivative which includes azides, the azide derivative was converted to the final product by the reaction of
L-glutamic acid in DMSO in the presence of TEA (
Scheme 35) [
78].
Scheme 35. Synthesis of 6R-Lometrexol starting from a double deprotected DDAH4Pte–OH 126.
In another approach, the lipase-catalyzed enantioselective esterification of 2-(4-bromophenethyl)propane-1,3-diol, derived in several steps from 2-(4-bromphenyl)acetic acid, was utilized in the asymmetric synthesis of key (
R)-2-amino-6-(4-bromophenethyl)-5,6,7,8-tetrahydropyrido[2,3-
d]pyrimidin-4(3
H)-one, which afforded the target product in two synthetic steps (Scheme 36) [
79,
80].
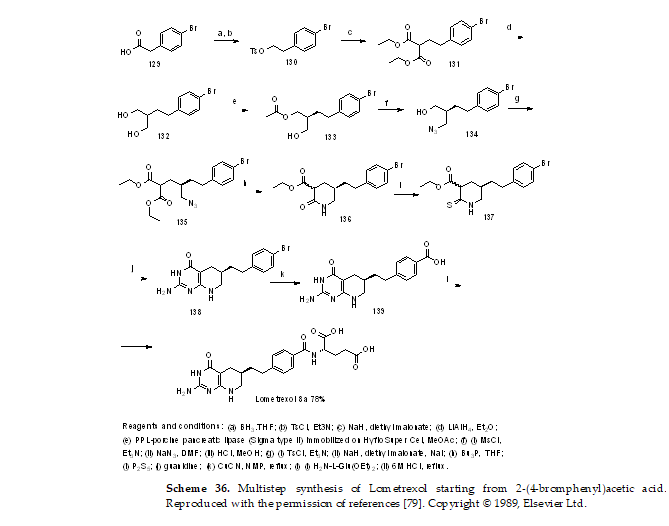
Scheme 36. Multistep synthesis of Lometrexol starting from 2-(4-bromphenyl)acetic acid. Reproduced with the permission of references [79]. Copyright © 1989, Elsevier Ltd.
3. Conclusions and Future Perspectives
In summary, this review represents the analysis of the most up-to-date synthetic ap- proaches for the synthesis of therapeutically significant analogues of folic acid, such as Lometrexol, Methotrexate, Pemetrexed, Pralatrexate, Raltitrexed, and TNP-351. Among the other folic acid analogues exhibiting antimalarial/antiprotozoal [81] and broad-spec- trum antimicrobial activity [82–84], the importance and effectiveness of the abovemen- tioned six analogues of folic acid as drugs or drug candidates for the treatment of diseases with a social significance, such as various types of cancers, severe psoriasis, and rheuma- toid arthritis, were reported in a large number of original research publications and re- view articles [18,30–32,40,51–55,67,69–71]. Even though folates were reported to be some- how connected with the severeness of COVID-19 [85–87], several recent studies suggested the effectiveness of antifolates for the therapy of patients with coronavirus SARS-CoV-2 [85,88,89], along with the enhancement of the antiviral efficacy of remdesivir [88], treat- ment of fungal infections with COVID-19-like symptoms [90], as well as treatment of fun- gal infections among COVID-19 patients [90].
Most of the synthetic strategies for these important scaffolds presented in research articles and patents are based on similar approaches and have only minor differences from each other. So far, no attention has been paid to methods based on transitional metal (TM)- catalyzation or the TM-free direct C-H-activation/С-H-functionalization of aza-aromatic
Scheme 36. Multistep synthesis of Lometrexol starting from 2-(4-bromphenyl)acetic acid. Reproduced with the permission of references [79]. Copyright © 1989, Elsevier Ltd.
3. Conclusions and Future Perspectives
In summary, this review represents the analysis of the most up-to-date synthetic ap- proaches for the synthesis of therapeutically significant analogues of folic acid, such as Lometrexol, Methotrexate, Pemetrexed, Pralatrexate, Raltitrexed, and TNP-351. Among the other folic acid analogues exhibiting antimalarial/antiprotozoal [81] and broad-spectrum antimicrobial activity [82–84], the importance and effectiveness of the abovementioned six analogues of folic acid as drugs or drug candidates for the treatment of diseases with a social significance, such as various types of cancers, severe psoriasis, and rheumatoid arthritis, were reported in a large number of original research publications and review articles [18,30–32,40,51–55,67,69–71]. Even though folates were reported to be somehow connected with the severeness of COVID-19 [85–87], several recent studies suggested the ef- fectiveness of antifolates for the therapy of patients with coronavirus SARS-CoV-2 [85,88,89], along with the enhancement of the antiviral efficacy of remdesivir [88], treatment of fungal infections with COVID-19-like symptoms [90], as well as treatment of fungal infections among COVID-19 patients [90].
Most of the synthetic strategies for these important scaffolds presented in research articles and patents are based on similar approaches and have only minor differences from each other. So far, no attention has been paid to methods based on transitional metal (TM)-catalyzation or the TM-free direct C-H-activation/C-H-functionalization of aza-aromatic rings as the most atom- and step-efficient approaches. We hope that our review will encourage future interest in this research area.
Author Contributions: Conceptualization, I.S.K., G.V.Z. and A.M.; methodology, I.S.K. and S.S.; software, S.S. and M.V.V.; validation, G.V.Z., A.M. and V.N.C.; formal analysis, G.V.Z., A.M. and V.N.C.; investigation, G.V.Z. and A.M.; resources, I.S.K. and G.V.Z.; data curation, I.S.K. and S.S.; writing—original draft preparation, I.S.K., G.V.Z. and S.S.; writing—review and editing, G.V.Z. and S.S.; visualization, A.M. and M.V.V.; supervision, G.V.Z., A.M. and V.N.C.; project administration, G.V.Z. and S.S.; funding acquisition, G.V.Z. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding: The analysis of synthetic approaches to antifolates was supported by the Russian Science Foundation (Project # 20-43-01004). The analysis of biological activity of antifolates was supported by the Ministry of Science and the Higher Education of RF (Ref. # 075-15-2022-1118, dated 29 June 2022).
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Data Availability Statement: Not applicable.
Conflicts of Interest: The authors declare no conflict of interest.