Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Dean Liu and Version 1 by Hyun-Min Kim.
CRISPR-Cas allows us to introduce desired genome editing, including mutations, epitopes, and deletions, with unprecedented efficiency. The development of CRISPR-Cas has progressed to such an extent that it is now applicable in various fields, with the help of model organisms. C. elegans is one of the pioneering animals in which numerous CRISPR-Cas strategies have been rapidly established over the past decade. Ironically, the emergence of numerous methods makes the choice of the correct method difficult. Choosing an appropriate selection or screening approach is the first step in planning a genome modification.
- CRISPR
- Cas
- genome editing
- C. elegans
1. Introduction
CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats-associated) is the current choice for genome editing. It is an RNA-guided system where a crRNA and a trRNA, together called a guide RNA (sgRNA), direct a Cas9 nuclease to the target gene of interest (Figure 1, [1]). The crRNA consists of a 20-nucleotide sequence from the spacer of the CRISPR locus and corresponds to a target DNA. The trRNA is complementary to a pre-crRNA, thus forming an RNA duplex later cleaved by RNase III to form a crRNA–trRNA hybrid, thereby directing the Cas9 RGN to make a double-stranded break (DSB) at the target site. The DSB can then be repaired by non-homologous end joining (NHEJ) or homologous recombination (HR)/homology-directed repair (HDR). In C. elegans, CRISPR-Cas9 technology was first adopted in 2013 (Figure 2), and since then, the nematode-research community has produced increasingly sophisticated strategies for genome editing. Here, wresearche rs introduce the significant discoveries of CRISPR-Cas genome editing achieved in the last decade (Figure 2).
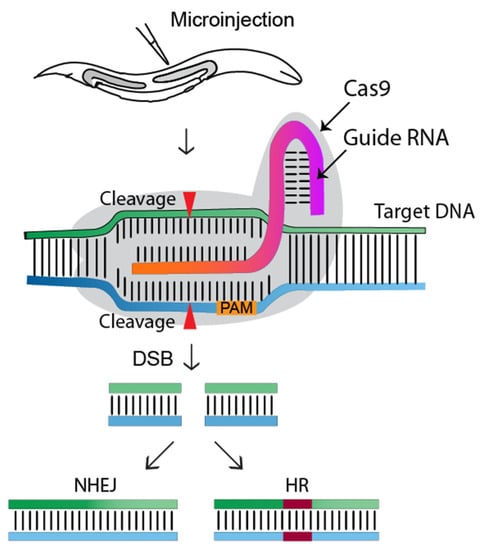
Figure 1. Overview of the CRISPR-Cas9 genome editing approach in C. elegans. The gonads of adult hermaphrodites are injected with the CRISPR-Cas9-containing DNA mixture. Injected Cas9 (grey color) forms a sequence-specific endonuclease when complexed with crRNA and trRNA (together called a guide RNA, or single guide RNA). The Cas9-guide RNA complex recognizes a target DNA sequence containing a PAM sequence (NGG) and induces a double-strand break (DSB), which will be repaired by non-homologous end joining (NHEJ) or homologous recombination (HR/HDR). Transgenic worms will be identified via marker-free (PCR) or marker-dependent strategies [1].
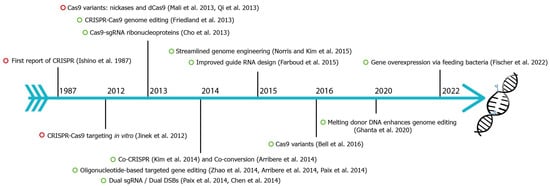
Figure 2. Fundamental discoveries and advances in C. elegans CRISPR-Cas genome editing. Timeline highlighting major events of C. elegans CRISPR-Cas9 (green circle) and other species (red circle). CRISPR were first identified in E. coli in 1987 [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21].
2. Cas9 Variants
Over the past decade, several CRISPR nuclease variants have been developed, and the range of its targets has been expanded (Figure 5). Nickases create a single-strand rather than a double-strand break. Targeting with nickase and two adjacent sgRNAs creates double-strand breaks with overhang, with reduces the off-target effects compared to the canonical/wild-type Cas9 system [4]. Dead Cas9 (dCas9) is an inactive catalytic nuclease. While the native Cas9 induces double-strand breaks on target DNA [5], dCas9 provides a binding site for activators or enhancers, promoting gene expression.
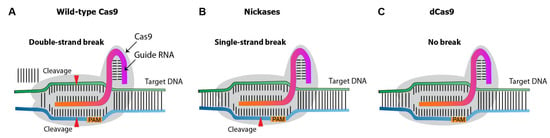
Figure 5. Diagram illustrating different types of Cas9 variants. (A), wild-type Cas9 nuclease. (B), Cas9 nickases that introduce a single-strand break at the target DNA. (C), catalytically inactive (dead) dCas9, which induces no breaks.
Since the C. elegans genome is an AT-rich genome, targeting conventional NGG near DSBs is frequently challenging. Therefore, having alternative sets of PAM sequences would expand the regions capable of being genome-edited. Kleinstiver et al. reported that Cas9 VQR (which possesses the amino acid substitutions D1135V, R1335Q, and T1337R) recognizes NGA PAM sequences instead of canonical NGG [24][22]. VQR recognizes NGAG as efficiently as wild-type Cas9 targets NGG [15], thus broadening the range of targets. Additionally, SpG and SpRY are two modified versions of Cas9, with more relaxed PAM requirements than Cas9 [25][23]. Thus, they target more portions of the genome. Of note, SpG and SpRY performed as efficiently as wild-type Cas9 at an increased concentration of CRISPR-Cas reagents (8 µM in the injection mix).
3. Guide RNA
Since guide RNA must designate the unique site of a gene of interest, multiple factors must be considered to achieve a high efficacy and specificity regarding genome editing. Studies have investigated strategies for designing guide RNA to enhance the genome editing frequency. For example, Farboud et al. reported that a GG motif at the 3′ end of the target sequences induced a high frequency of mutagenesis via NHEJ and HR pathways [16]. The median frequency at the targets was 10-fold higher than those reported in previous studies with multiple guide RNAs [6,11,26][6][11][24].
Later, the same group explored the location of DSB repair events in CRISPR-Cas editing. Interestingly, NHEJ induces asymmetric insertions and deletions (indels) preferentially in regions of 5′ of PAM [27][25]. Notably, a similar propensity for repair has been found in mammals, advocating the use of C. elegans as a model to understand the universal rules underlying genome editing [28,29][26][27].
Since variants of Cas nucleases have expanded the range of genome targets, identifying a proper target may now involve additional effort. Currently, a web-based database of guide RNA can help to identify guide RNAs for target genes and minimize off-target sites. Several database services can identify potential guide RNA for the entire C. elegans genome (genome.sfu.ca/gexplore (accessed on 4 December 2022), www.crisprscan.org (accessed on 4 December 2022), crispr.dbcls.jp (accessed on 4 December 2022), crispor.tefor.net (accessed on 4 December 2022) [30,31,32,33][28][29][30][31]). The user can search the guides present in the database by entering multiple factors, including genomic interval, GC content, gene name, and the presence of GG at the 3′ end of the guides. Notably, CRISPRscan can identify targets for the Cas9 variants SpG and SpRY.
4. Dual sgRNA/Dual DSBs
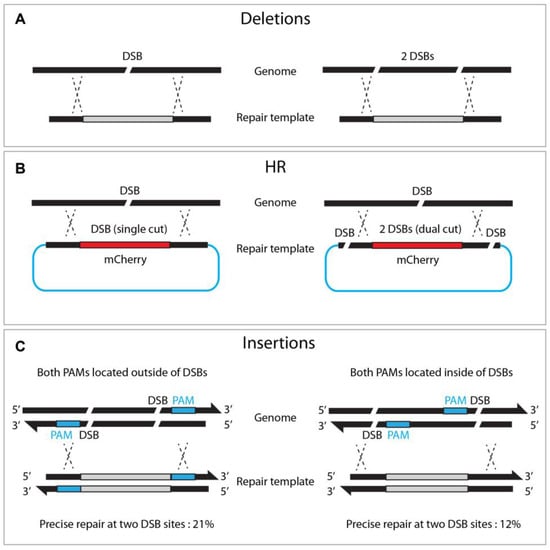
While Cas9 nuclease induces a single DSB at a target site, dual or triple DSBs offer advantages for genome editing. For example, deletion mutants can be achieved by adopting two sgRNAs via NHEJ or HR pathways (Figure 6A, [17]). Likewise, dual sgRNA can generate long chromosome deletion between two sgRNAs [18]. Chen et al. also demonstrated that dual DSBs generate reciprocal chromosomal translocation, thereby providing a practical approach to studying genome rearrangement [34][32].
Figure 6. Dual DSBs enhance deletions, homologous recombination, and insertions. (A) Single versus dual DSBs at targeted sites. (B) Dual DSBs (right) in the repair template enhances HR efficacy compared to single DSB (left). (C) Locating both PAMs outward of the DSB sites enhances insertion frequency.
Zhang et al. reported that dual cutting of the repair template, rather than genomic DNA, enhanced precise genome editing efficacy (Figure 6). The dual-cut repair template, flanked by two sgRNA at both ends of the plasmid, increased HR efficiency by between two and fivefold compared to a conventional uncut circular template. They observed a proportional increase in HR efficiency, with a more extended homologous arm in either circular or linear DNA, suggesting that a longer flanking sequence improves dual-cut-mediated HR efficacy [35][33]. Similarly, dual DSBs promoted the insertion of large (9300 bp) DNA fragments, in combination with a dpy-10 co-conversion strategy, where single DSBs failed (5% vs. 0%, respectively) [27][25]. It is worth noting that, when the dsDNA template was cleaved by two DSBs, orienting both PAMs outward resulted in efficient homologous recombination (21% vs. 12% in an out/out and in/in configuration, respectively, Figure 6).
5. Oligonucleotide as a Repair Template for Homologous Recombination
Since linear DNA is prone to degradation by nucleases present in the cells, the conventional way of delivering a repair template is to supply double-stranded DNA as a part of circular DNA. Therefore, plasmid DNA is desirable, especially for large-sized repair templates. However, a growing number of studies have found that single-stranded oligonucleotides can often be good alternatives for HR-mediated genome editing. Specifically, oligonucleotides offer a few advantages over plasmid DNA: they are cloning-free and can be rapidly synthesized via commercially available resources. Therefore, adopting linear DNA reduces the amount of time required for the entire genome editing process.
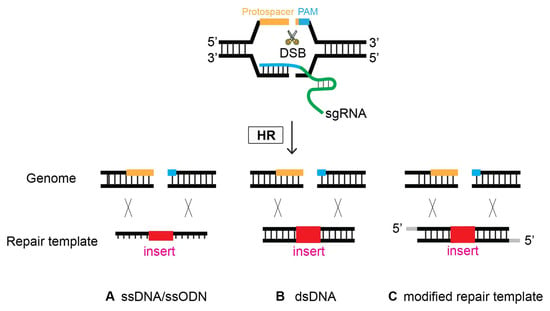
The first demonstration of oligonucleotides as repair templates was performed in 2014, substantiating their simplicity and efficacy [19]. An oligonucleotide, ~100 bp long, serves as a template to repair DSBs occurring at four different genes via homologous recombination (Figure 7). This strategy has demonstrated its efficiency in several labs [12,36,37][12][34][35]. It is worth noting, however, that it appears that smaller flanking homology requires the DSB/cleavage site to be near the site of genome editing [12].
Figure 7. Oligonucleotides can serve as a template to repair DSBs via HR. At the site of CRISPR-Cas9-induced DSB, Single-stranded DNA (ssDNA), double-stranded DNA (dsDNA), and modified repair templates serve as a repair template. (A) ssDNA, (B) dsDNA, and (C) modified nucleotides as a repair template.
6. Modified Repair Template
Previous research investigated whether modification of the donor template improves HR efficacy. Although controversial at first, a growing number of studies have reported that the modification of repair donors enhances genome editing proficiency (Figure 7). In 2014, Zhao et al. employed a phosphorothioate-modified oligonucleotide as a repair template in C. elegans for the first time. Although the modified oligonucleotide resulted in the desired genome editing, it was unclear whether its efficacy was enhanced compared that resulting from to non-modification [19]. In mammalian cultured cells, however, phosphorothioate-modified oligonucleotides enhanced the genome editing efficiency of single-stranded oligonucleotide donors. In addition, modified oligonucleotides allow for insertions > 100 bp long, providing design flexibility [37][35]. In contrast, fluorescent and amine modifications to the 5′- and 3′-termini of single-stranded oligodeoxynucleotide (ssODN) donors did not alter HR frequency compared to nonmodified donors in human cells [38][36].
With this controversy, recent studies have explored donors with 5′-end modifications that enhance genome editing efficacy. Gutierrez-Triana et al. showed that adding biotin at the 5′ ends of dsDNA leads to an increase in HR efficiency of up to 60% in the injected generation of medaka fish embryos. Provocatively, the authors demonstrated that biotin and SpC3 5′ modifications prevent donor multimerization/NHEJ of dsDNA, thereby providing optimal conditions for HR-mediated CRISPR-Cas genome editing [39][37]. Similarly, Yu et al. reported that 5′ C6-PEG10-modified dsDNA increased knockin frequency up to fivefold, in combination with Cas9 ribonucleoprotein (RNP) in human cells [40][38]. In line with these reports in fish and mammalian cells, the C. elegans study demonstrated that 5′ modifications of the donor improved the efficacy of HR frequency roughly twofold [41][39]. Interestingly, TEG (triethylene glycol) and RNA::TEG modifications performed with similar efficacy in C. elegans, whereas RNA::TEG was superior to TEG in human cells and zebrafish. They demonstrated that these 5′ modifications suppress donor ligation reactions in a similar way to that of biotin and SpC3 5′ modifications [39][37].
It is worth noting that C. elegans studies from the Mello Lab reported additional alterations that enhance HR efficiency dramatically by modifying repair template DNA. First, a single-stranded overhang containing dsDNA donors yielded higher integration rates at three loci [36][34]. Second, denaturing and cooling the dsDNA donor template increased the HR frequency by up to 50% [20].
References
- Kim, H.M.; Colaiacovo, M.P. CRISPR-Cas9-Guided Genome Engineering in Caenorhabditis elegans. Curr. Protoc. Mol. Biol. 2019, 129, e106.
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821.
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838.
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183.
- Friedland, A.E.; Tzur, Y.B.; Esvelt, K.M.; Colaiacovo, M.P.; Church, G.M.; Calarco, J.A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 2013, 10, 741–743.
- Lo, T.W.; Pickle, C.S.; Lin, S.; Ralston, E.J.; Gurling, M.; Schartner, C.M.; Bian, Q.; Doudna, J.A.; Meyer, B.J. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics 2013, 195, 331–348.
- Chen, C.; Fenk, L.A.; de Bono, M. Efficient genome editing in Caenorhabditis elegans by CRISPR-targeted homologous recombination. Nucleic Acids Res. 2013, 41, e193.
- Tzur, Y.B.; Friedland, A.E.; Nadarajan, S.; Church, G.M.; Calarco, J.A.; Colaiacovo, M.P. Heritable custom genomic modifications in Caenorhabditis elegans via a CRISPR-Cas9 system. Genetics 2013, 195, 1181–1185.
- Cho, S.W.; Lee, J.; Carroll, D.; Kim, J.S.; Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics 2013, 195, 1177–1180.
- Kim, H.; Ishidate, T.; Ghanta, K.S.; Seth, M.; Conte, D., Jr.; Shirayama, M.; Mello, C.C. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics 2014, 197, 1069–1080.
- Arribere, J.A.; Bell, R.T.; Fu, B.X.; Artiles, K.L.; Hartman, P.S.; Fire, A.Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 2014, 198, 837–846.
- Norris, A.D.; Kim, H.M.; Colaiacovo, M.P.; Calarco, J.A. Efficient Genome Editing in Caenorhabditis elegans with a Toolkit of Dual-Marker Selection Cassettes. Genetics 2015, 201, 449–458.
- Dickinson, D.J.; Pani, A.M.; Heppert, J.K.; Higgins, C.D.; Goldstein, B. Streamlined Genome Engineering with a Self-Excising Drug Selection Cassette. Genetics 2015, 200, 1035–1049.
- Bell, R.T.; Fu, B.X.; Fire, A.Z. Cas9 Variants Expand the Target Repertoire in Caenorhabditis elegans. Genetics 2016, 202, 381–388.
- Farboud, B.; Meyer, B.J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics 2015, 199, 959–971.
- Paix, A.; Wang, Y.; Smith, H.E.; Lee, C.Y.; Calidas, D.; Lu, T.; Smith, J.; Schmidt, H.; Krause, M.W.; Seydoux, G. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 Sites in Caenorhabditis elegans. Genetics 2014, 198, 1347–1356.
- Chen, X.; Xu, F.; Zhu, C.; Ji, J.; Zhou, X.; Feng, X.; Guang, S. Dual sgRNA-directed gene knockout using CRISPR/Cas9 technology in Caenorhabditis elegans. Sci. Rep. 2014, 4, 7581.
- Zhao, P.; Zhang, Z.; Ke, H.; Yue, Y.; Xue, D. Oligonucleotide-based targeted gene editing in C. elegans via the CRISPR/Cas9 system. Cell Res. 2014, 24, 247–250.
- Ghanta, K.S.; Mello, C.C. Melting dsDNA Donor Molecules Greatly Improves Precision Genome Editing in Caenorhabditis elegans. Genetics 2020, 216, 643–650.
- Fischer, F.; Benner, C.; Goyala, A.; Grigolon, G.; Vitiello, D.; Wu, J.; Zarse, K.; Ewald, C.Y.; Ristow, M. Ingestion of single guide RNAs induces gene overexpression and extends lifespan in Caenorhabditis elegans via CRISPR activation. J. Biol. Chem. 2022, 298, 102085.
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.R.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485.
- Vicencio, J.; Sánchez-Bolaños, C.; Moreno-Sánchez, I.; Brena, D.; Vejnar, C.E.; Kukhtar, D.; Ruiz-López, M.; Cots-Ponjoan, M.; Rubio, A.; Melero, N.R.; et al. Genome editing in animals with minimal PAM CRISPR-Cas9 enzymes. Nat. Commun. 2022, 13, 2601.
- Waaijers, S.; Portegijs, V.; Kerver, J.; Lemmens, B.B.; Tijsterman, M.; van den Heuvel, S.; Boxem, M. CRISPR/Cas9-targeted mutagenesis in Caenorhabditis elegans. Genetics 2013, 195, 1187–1191.
- Farboud, B.; Severson, A.F.; Meyer, B.J. Strategies for Efficient Genome Editing Using CRISPR-Cas9. Genetics 2019, 211, 431–457.
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826.
- Richardson, C.D.; Kazane, K.R.; Feng, S.J.; Zelin, E.; Bray, N.L.; Schafer, A.J.; Floor, S.N.; Corn, J.E. CRISPR-Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat. Genet. 2018, 50, 1132–1139.
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 2015, 12, 982–988.
- Naito, Y.; Hino, K.; Bono, H.; Ui-Tei, K. CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 2015, 31, 1120–1123.
- Concordet, P.J.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245.
- Hutter, H.; Ng, M.P.; Chen, N. GExplore: A web server for integrated queries of protein domains, gene expression and mutant phenotypes. BMC Genom. 2009, 10, 529.
- Chen, X.; Li, M.; Feng, X.; Guang, S. Targeted Chromosomal Translocations and Essential Gene Knockout Using CRISPR/Cas9 Technology in Caenorhabditis elegans. Genetics 2015, 201, 1295–1306.
- Zhang, J.P.; Li, X.L.; Li, G.H.; Chen, W.; Arakaki, C.; Botimer, G.D.; Baylink, D.; Zhang, L.; Wen, W.; Fu, Y.W.; et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017, 18, 35.
- Dokshin, G.A.; Ghanta, K.S.; Piscopo, K.M.; Mello, C.C. Robust Genome Editing with Short Single-Stranded and Long, Partially Single-Stranded DNA Donors in Caenorhabditis elegans. Genetics 2018, 210, 781–787.
- Renaud, J.B.; Boix, C.; Charpentier, M.; De Cian, A.; Cochennec, J.; Duvernois-Berthet, E.; Perrouault, L.; Tesson, L.; Edouard, J.; Thinard, R.; et al. Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell. Rep. 2016, 14, 2263–2272.
- Lee, K.; Mackley, V.A.; Rao, A.; Chong, A.T.; Dewitt, M.A.; Corn, J.E.; Murthy, N. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. Elife 2017, 6, e25312.
- Gutierrez-Triana, J.A.; Tavhelidse, T.; Thumberger, T.; Thomas, I.; Wittbrodt, B.; Kellner, T.; Anlas, K.; Tsingos, E.; Wittbrodt, J. Efficient single-copy HDR by 5′ modified long dsDNA donors. Elife 2018, 7, e39468.
- Yu, Y.; Guo, Y.; Tian, Q.; Lan, Y.; Yeh, H.; Zhang, M.; Tasan, I.; Jain, S.; Zhao, H. An efficient gene knock-in strategy using 5’-modified double-stranded DNA donors with short homology arms. Nat. Chem. Biol. 2020, 16, 387–390.
- Ghanta, K.S.; Chen, Z.; Mir, A.; Dokshin, G.A.; Krishnamurthy, P.M.; Yoon, Y.; Gallant, J.; Xu, P.; Zhang, X.-O.; Ozturk, A.R.; et al. 5′-Modifications improve potency and efficacy of DNA donors for precision genome editing. eLife 2021, 10, e72216.
More