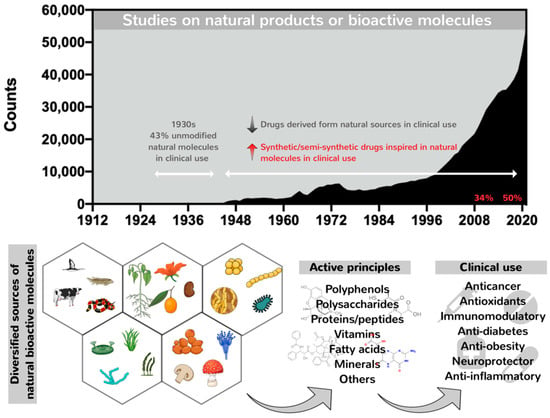
Nature has long proved an outstanding source of bioactive molecules with vast structural complexity and diversity. These have been used for millennia, mainly as component mixtures in crude extracts, as a unique strategy to treat several physiopathological conditions, including wounds, infectious diseases and other disorders
[1][2][1,2]. In contrast, conventional drugs approved for therapeutic use are mainly represented by synthetic and small molecules ranging from 900 to 1500 Da designed and chemically synthetized to fit a pre-determined target via high-throughput screening. However, since the targets for small therapeutic molecules represent only 2–5% of the human genome products, the search for alternative therapeutics capable of expanding the number of new targets has increased the number of studies on natural therapeutic biomolecules, comprising searches in diverse classes, including natural macromolecules particularly proteins, peptides and polysaccharides
[3][4][3,4]. The first efforts to identify and isolate active principles from natural extracts began in the 19th century with the isolation of the anti-malarial quinine, the opiate analgesic morphine and salicylic acid, creating the medicinal use of naturally isolated compounds. Since then, many other bioactive compounds have been isolated, mainly alkaloids, such as caffeine, nicotine, codeine, atropine, colchicine, cocaine and capsaicin
[5]. The utilization of unmodified natural molecules increased by 43% during the 1930s (
Figure 1). After this period, a decline in the number of unmodified natural products entering clinical practices took place, although the number of published data on natural products and bioactive molecules has increased. The past 30 years have been marked by a profile shift characterized by the replacement of unmodified natural molecules by semisynthetic and synthetic derivatives chemically designed but inspired by natural molecules (
Figure 1)
[6]. This profile shift was stimulated by the emergence of modern high-throughput platforms for screening and synthetic combinatorial strategies aimed at the fast discovery of new drug candidates similar to or analogs of natural molecules displaying special activities identified over the years, conducted through semi-synthetic modifications or by total synthesis, but still maintaining their pharmacophore. Moreover, the generation of synthetic/semisynthetic analogs is in contrast to time-consuming and high-cost methodologies applied to the identification of natural compounds in crude extracts, followed by their isolation and obtention in bulk amounts, impacting the emergence of new but unmodified natural pharmacological compounds for clinical use
[7][8][9][7,8,9]. Because of this, the chemical synthesis of natural products was prioritized to optimize pharmaceutical production, improving the purity, quality and yields of bioactive compounds with reduced costs. Salicylic acid was the first natural product synthesized in 1853, copying the natural molecule
[5].
2. Evolution of Drug Delivery Systems and the Emergence of Nanotechnology in Clinical Treatments
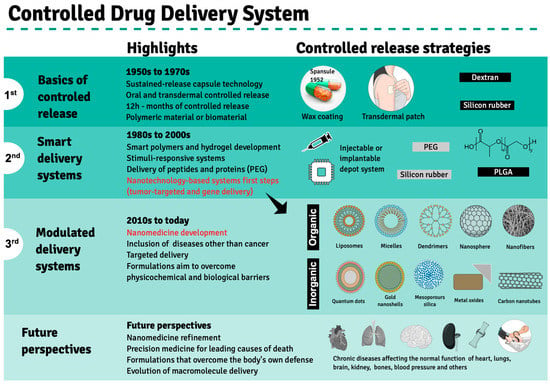
Drug performance depends primarily on the type of delivery and release rate in order to guarantee a sustained drug amount that matches the therapeutic index between the maximum safe concentration and above the minimum effectiveness in the blood stream, avoiding abrupt peak concentrations as a result of massive drug release or the need for multiple doses to achieve drug effectiveness. These limitations encouraged the search for an ideal drug delivery system to substitute pills and capsules that release active compounds in aqueous media, termed immediate release (IR) formulations. The pioneer strategy in this regard comprised Spansules
®, a first-generation drug delivery system, where micro-pellets were coated with a water-soluble wax of varied thicknesses, allowing for the controlled release of oral drugs for up to 12 h, leading to a constant concentration in blood. The first-generation drug delivery period, which ran from the 1950s to the 1970s, was also marked by the development of silicon- or dextran-based transdermal formulations based on controlled release, mainly through dissolution and diffusion (
Figure 2). Both delivery systems faced several challenges, leading to advanced-generation systems
[20][21].
Figure 2. Development of controlled drug delivery systems. Three distinct development phases—1st, 2nd and 3rd generations—are highlighted based on therapeutic products approved by the FDA since the 1950s, when Spansule
®, the first drug release system, was approved for clinical use. The main highlights of each phase are depicted in the central column, while the main synthetic or biological material used in the drug delivery system preparation is indicated in the left column. The advent of nanotechnological drug delivery strategies is highlighted in red during the last decade of the second development phase. Future perspectives are exhibited at the bottom panel. Created using Mind the Graph (
https://mindthegraph.com accessed on 14 August 2022).
The second generation of drug delivery formulations began in 1980 and lasted until 2010 and came with the understanding that it is not indispensable that drug concentrations be maintained at fixed levels. This period was marked by the development of smart polymers and hydrogels that evolved alongside biodegradable microparticles, solid implants and in situ gel-forming implants capable of delivering long-term release and stimuli-responsive bioactive compounds. Finally, nano-delivery systems emerged, where nanoparticles obtained from biodegradable polymers, polymeric micelles, lipids, chitosan and dendrimers are used to carry anticancer agents and gene sequences. The need to overcome biological and physicochemical barriers gave rise to the third generation of drug delivery systems, in which advanced nanomaterials enable the delivery of poorly water-soluble and/or very labile drugs or cell components, including peptides, proteins and DNA or RNA sequences. Novel delivery concepts include targeted drug delivery using nanoparticles and self-regulated drug delivery
[20][21]. However, few drugs were approved by the FDA, even though much experimental evidence has proven that drug delivery employing nanotechnology systems could enhance the effectiveness of anticancer drugs against tumors in animal models
[10].
According to the National Nanotechnology Initiative (2021), nanotechnology involves the manipulation of nanoparticles ranging from 1 to 100 nm, and their use is widespread in several areas, including engineering, physics and informatics. However, nanotechnology applications for pharmaceutical purposes may cause the most significant impact on human health and, because of this, has been considered the most promissory technology for treatments against degenerative pathologies such as cancer
[21][22][22,23] and central nervous system disorders
[23][24][24,25], as well as in antiviral therapy to aid in SARS-CoV-2 immunization and treatment
[25][26][26,27].
The morphology of nano-scaled particles led to the optimization of treatments by enabling nano-materials to reach physiological sites that used to remain inaccessible, such as specific areas of the brain damaged by synucleinopathies or brain neoplasms, which require the ability to cross the blood–brain barrier to achieve therapeutic intervention success
[24][25]. Moreover, nanomaterials exhibit a large surface/volume ratio, potentiating drug effects on target sites (cell, tissue or organ), comprising efficient bioactive molecule nanocarriers that can be encapsulated through adsorption, core entrapment or covalent surface binding. The nano-encapsulated bioactive molecule is released constantly and in a controlled manner, reducing adequate drug dosages to achieve pharmacological effects and minimize side effects widely attributed to conventional pharmaceuticals
[27][28][29][28,29,30].
Nano-encapsulated compounds can be protected from degradation in the bloodstream. They can still reach intracellular compartments via endosomes through passive permeability, releasing bioactive compounds into the cytoplasm or directing them to intracellular targets with ligands associated with nano-capsule internalization
[30][31]. In anti-tumorigenic therapies, the passive accumulation of anticancer drugs at the tumor localization can be increased by facilitating the permeability and retention effect (EPR), a phenomenon following the local inflammatory status of tumor blood vessels that become leaky, allowing for the passage and accumulation of nanometric materials in tumor tissues
[31][32][33][34][32,33,34,35].
The primary chemical composition of nanoparticles described for medicinal therapy treatments comprises organic or inorganic compounds. The former includes natural or synthetic polymers such as chitosan, collagen, glycerol, polylactic-co-glycolic acid (PLGA) and dendrimers, as well as lipid-based materials, such as liposomes and micelles. At the same time, inorganic nanoparticles comprise gold nano-shells, metal oxides, carbon nanotubes and quantum dots (
Figure 2)
[35][36]. Molecules for therapeutic purposes can be encapsulated by these nanoparticles or complexed to them by adsorption or covalent binding to the nanocarrier surface. Depending on the carried bioactive compound, these nanoconjugates may be used to treat several pathologies, including degenerative diseases such as atherosclerosis and Parkinson’s disease, as well as cancers, as mentioned previously (
Table 1).
Table 1. Pre-clinical studies on nano-delivery systems demonstrated in cell cultures and animal models for the loading of therapeutics aiming at the treatment of several health disorders.
PD—Parkinson’s disease.
The application of hydrophobically modified chitosan nanoparticles in the delivery of silibinin, a flavo-lignan isolated from the seeds of the milk thistle plant, was shown to improve the response promoted by a sustained release and enhance the solubility of this poorly aqueous soluble compound
[46][47]. Similarly, resveratrol (RV), a polyphenol non-flavonoid commonly found in red or dark grapes, encapsulated in albumin nanoparticles (NPs) and functionalized with the tripeptide arginine-glycine-aspartate (RGD), demonstrated a prolonged RV blood circulation time and increased content surrounding a target tumor by 8.1-fold compared to free-RV. In a murine model, RV-NPs-RGD suppressed tumor growth with no relapse after 35 days of treatment, while progressive tumor growth was observed in an RV-free treatment, indicating that RV-NPs-RGD should be considered a promising chemotherapy agent
[47][48][48,49].
Curcumin, another widely studied natural antioxidant and anticancer compound derived from turmeric or saffron, displays limited clinical application due to its molecule instability and poor solubility. However, curcumin encapsulated in solid lipid nanoparticles displays a high antiproliferative effect against SKBR3 cancer cells compared to free curcumin, with nanoencapsulation improving this compound’s bioavailability. Moreover, nano-encapsulated curcumin induced high-extension SKBR3 cell apoptosis (36.7%) and pronounced cell migration inhibition
[49][50].
The co-encapsulation of tocotrienol (TRF) and simvastatin (SIM) in lipid nanoparticles has been reported as displaying an anti-proliferative effect on the breast adenocarcinoma +SA cell line, with an IC
50 of 0.52 μM, when compared to each pharmaceutical separately. Nearly 25 to 40% of SIM is released over 48 h, with a quicker release in the first 10 h, followed by a slower and controlled kinetic release throughout the following 38 h
[36][37]. Similarly, when lianol (LN) was encapsulated in solid nanoparticles, it affected HepG2 hepatocarcinoma and A549 pulmonary adenocarcinoma cell lines
[37][38].
Folic acid conjugated through ionic interactions with chitosan nanoparticles (FA-CS) loaded with vincristine induces apoptosis in 75% of NCI-H460 lung cancer cells, in contrast to unloaded nanoparticles (31%)
[39][40]. Nanoencapsulation efficiency can be increased when prepared at 4:25 (
v/
v) of vincristine/FA-CS, reaching a 95% efficiency and loading capacity of 48.65%, reinforcing promising anticancer effects on tumorigenic lung cells
[39][40]. Similar effects were observed using PLGA (poly lactic-co-glycolic acid) nanofibers coated with metformin (MET), which exhibited cytotoxicity against A549 lung cancer cells after 48 h compared to free MET
[42][43].
Liposome encapsulation improves the pharmacological effects of bioactive molecules, including metallic complexes, contributing to toxicity constraints by decreasing effective doses. Encapsulation of iridium III complexes (Ir-1, Ir-2 and Ir-3) in nano-liposomes, for example, was shown to improve their anticancer activity against several human carcinoma lineages, such as HepG2, hepatocellular carcinoma; HTC-116, colon cancer; HeLa, cervical cancer; A549, lung carcinoma; BEL-7402, hepatocellular carcinoma; SGC- 7901, gastric adenocarcinoma; and Eca-109, esophagus cancer cell, and against B16 mouse melanoma cells but no toxicity against healthy nih3T3 murine cells. On the other hand, no activity was observed for free Ir-2, and the nano-liposomal formulation exhibited a superior IC50 compared to cisplatin, especially against the A549 cell line. The intraperitoneal administration of the nano-liposomal formulation loaded with the Ir-2 complex for 10 consecutive days prior to A549 carcinogenic cell inoculation in mice resulted in 57.45% tumor mass reduction. In vitro assays have shown that encapsulated complexes stimulate apoptotic activity induced by increased intracellular ROS and cell cycle arrest at G0/G1 phases
[40][41]. Similarly, TPGS-coated liposomes with SiRNA-corona Bcl-2 loaded with doxorubicin (Dox) promoted a 7-fold reduction in mice tumoral mass compared to free Dox
[41][42]. Dendrimer nanoparticles of poly(amidoamine) (PAMAM) have also been successfully used to deliver methotrexate and D-glucose (PAMAM-MTX-GLU), inhibiting MDA-MB 231 breast cancer cells
[43][44].
In addition to their use in anti-tumorigenic therapy, nanoparticles can be applied to treat other pathologies, including atherosclerosis and Parkinson’s disease. Mannose-functionalized dendrimer nanoparticles (mDNP) have been formulated to selectively deliver LXR-L, the liver receptor ligand, to macrophages associated with atherosclerotic plaque
[44][45]. Four weeks of mDNP-LXR-L administration in LDL-receptor knockout mice led to nearly a 10% reduction in atherosclerotic plaque, necrosis area and inflammatory response evaluated by the expression of the metalloproteinase 9 (MMP-9) gene, regulated by NF-kB, the nuclear factor kappa B. In contrast, no increases in the expression of hepatic lipogenic or plasma lipid genes were observed. Gold nanoparticles (AuNCs) containing N-isobutyryl-L-cysteine (L-NIBC) are promising in the treatment of Parkinson’s disease. An in vitro assay of the effect of 1-methyl-4-phenyliridine (MPP) on SH-SY5Y cells showed that the nanoparticles inhibited synuclein fiber aggregation and formation in the brain while also preventing the formation of Lewy bodies and the death and dysfunction of neurons, all histological characteristics observed in Parkinson’s disease
[45][46]. These data corroborated results described in mice pre-inoculated with the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and treated with AuNCs, which promoted the regression of behavioral disorders determined by the open field, swimming and rotarod tests, reinforcing the potential of nanoparticles loaded with L-NIBC to treat Parkinson’s disease.
The therapeutic efficacy of several pharmaceuticals has been improved by nanoparticles displaying the high potential of drugs complexed to nanomaterials to treat neoplasms, neurodegeneration and atherosclerosis, among other physiopathological conditions (
Table 1). However, despite the significant science and technology advances in obtaining nanocarriers for pharmaceuticals, the time required for optimization and regulation aiming at their commercialization can take at least thirteen years, without considering pre-clinical studies and large-scale production, as well as phase 0, I, II and III clinical trials that must precede the submission and approval of health regulatory agencies to finally reach phase IV, as defined by the American Cancer Society
[50][51][51,52].
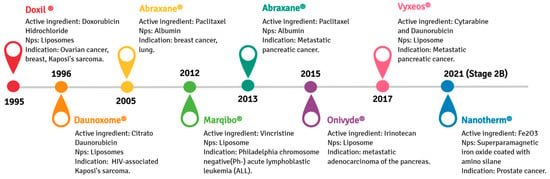
A nano-liposomal formulation of doxorubicin (DOX), Doxil
®, was the first drug- functionalized nanoparticle formulation approved by the U.S. Food and Drug Administration (FDA) in 1995. Doxil
® can accumulate in solid tumors due to the EPR phenomena, effectively treating several cancers, including metastatic ovarian cancer and AIDS-related Kaposi’s sarcoma
[52][53][54][53,54,55]. The anti-tumorigenic effect of Doxil
® is attributed to its DNA interleaving ability and topoisomerase II inhibition, resulting in the downregulation of DNA replication and RNA transcription. Doxil
® is not readily cleared from plasma by the mononuclear phagocytic system (MPS), which allows the continuous release of encapsulated DOX, improving its therapeutic performance compared to free-DOX
[55][56][56,57]. Another available chemotherapy formulation is Abraxane
®, in which paclitaxel is efficiently encapsulated in albumin nanoparticles. Abraxane
®, approved by the FDA in 2005 and originally used to treat breast cancer, was expanded to treat advanced pancreatic carcinoma in 2013
[57][58]. Its formulation improves paclitaxel bioavailability, resulting in higher intra-tumoral paclitaxel concentrations facilitated by endothelial transcytosis through the albumin receptor-mediated (gp60)
[58][59]. Other examples of nanostructured drugs developed for cancer therapy or diagnosis are depicted in
Figure 3.
Figure 3. Timeline from 1995 to 2021 concerning representative anticancer pharmaceuticals loaded in nanoparticles approved by the American Food and Drug Administration for clinical purposes.
3. Liposomal Formulation Performance: Characteristics, Functionalization and Internalization
An effective liposomal formulation can be achieved by choosing the suitable lipid composition, proper functionalization and a smart targeting strategy. Phospholipid selection, considering the head group and chain length and number of liposome components, are crucial in determining liposome safety, stability and efficiency
[59][94]. Besides their chemical composition, liposome efficiency depends on their stiffness, surface charge and lipid organization, as surface modifications interfere with liposome stability and can be handled to expand its use
[60][61][95,96].
The main liposome components are glycerophospholipids, amphiphilic lipids composed of a glycerol molecule linked to a phosphate group and two fatty acid chains that can be either saturated or unsaturated. Phosphatidylcholine and phosphatidylethanolamine, abundant in plants and animals, are the most employed to form liposomes
[62][63][97,98]. Liposomes can acquire positive, negative or neutral charges depending on the phospholipid head and chain, which confer their overall characteristics and functionalities
[64][65][99,100]. Stability can be conferred to liposomes formed by phospholipids with longer tails and low degrees of unsaturation and ether linkages. Phospholipids with longer saturated hydrocarbon chains display a remarkable ability to interact with each other and form tightly ordered bilayer structures. On the other hand, shorter unsaturated hydrocarbon chains form liposomes with fluid and disordered bilayers
[59][66][94,101].
Synthetic phospholipids can be formed by the modification of the head groups, aliphatic chains and alcohols of natural phospholipids, creating an enormous variety of synthetic phospholipids, such as 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dioleoyl-glycero- 3-phosphocholine (DOPC), 1,2-distearoyl-sn-glycero-3-phosphoglycerol (DSPG), 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol (DPPG), 1,2-dioleoyl-sn-glycerol -3-phosphoethanolamine, (DOPE) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), which have proven to be more stable
[67][102].
Liposomes are formed by the hydrophilic interactions between polar head groups, the van der Waals forces between hydrocarbon chains, holding the long hydrocarbon tails together, and hydrogen linkages with H
2O. In turn, H
2O repels the hydrophobic chains, and the liposomes self-assemble spontaneously into a closed bilayer
[67][68][102,103].
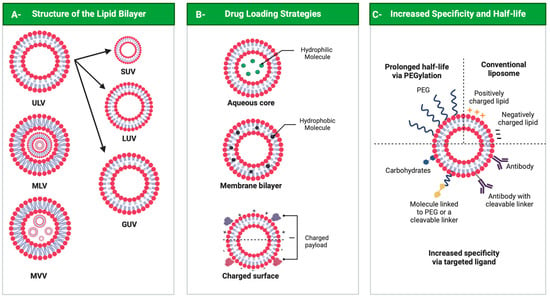
Liposomes can be classified as neutral, anionic and cationic according to lipid bilayer components. They can assume different 3D structures, as unilamellar vesicles (ULVs), multilamellar vesicles (MLVs) or multivesicular vesicles (MVVs). ULVs contain a single-lipid-bilayer membrane and can vary in size, with small unilamellar vesicles (SUVs) measuring between 30 and 100 nm, large unilamellar vesicles (LUVs) from 100 to 300 nm and giant unilamellar vesicles (GUVs) from 1 to 100 μm. In multilamellar vesicles, layers are concentric, while in MVVs, several smaller vesicles encase the interior of another vesicle (
Figure 4, panel A)
[69][70][104,105]. Another liposome formed by two bilayer membranes, the double-layer vesicle, comprises a framework that can improve liposome stability, which delays and sustains the release of their load
[71][106].
Figure 4. Liposomes. Panel A: Structure of liposomes. ULV—unilamellar vesicle (small ULV (SUV), large ULV (LUV) and giant ULV (GUV)); MLV—multilamellar vesicle; MVV—multivesicular vesicle. Panel B: Drug loading strategies. Panel C: Liposome surface and functionalization. All figures were drawn with BioRender accessed on 1 December 2022.
In addition to phospholipids, its main components, cholesterol, glycol-derivatives including propylene glycol and polyethylene glycol (PEG) and even polymers such as chitosan, can increase liposome stability. These components can promote pronounced effects on healthy tissues and cells and activate or suppress immune responses
[72][73][107,108]. Cholesterol incorporation into liposome bilayers can influence their fluidity, reducing their permeability and increasing in vitro and in vivo stabilities. Cholesterol, a hydrophobic molecule, induces a dense phospholipid packing and inhibits interactions among lipid chains interspersed between them, promoting liposome membrane stabilization
[74][75][76][109,110,111]. The cholesterol molecule accommodates its hydroxyl group close to the hydrophilic region of phospholipids, and its aromatic ring lays parallel to the fatty acid chains in the lipid bilayer
[77][112]. In the absence of cholesterol, liposomes can interact with proteins such as albumin, transferrin, macroglobulin and high-density lipoproteins (HDL), destabilizing the liposomal membrane structure and, consequently, decreasing drug delivery system performance
[78][79][80][113,114,115].
Polymers such as chitosan are also used for liposomal surface modification, leading to a protective shell on the liposome surface, mainly for oral drug delivery
[73][81][108,116]. Glycols such as propylene glycol incorporated into phospholipid vesicles with polyethylene glycol (PEG) have been advocated as flexible lipid vesicles to improve drug delivery systems targeting the skin
[82][83][117,118]. Different PEGs on liposome surfaces can prolong their half-lives in the bloodstream, from a few minutes considering conventional liposomes to several hours for stealth liposomes, also called PEGylated liposomes
[77][112]. One of the main disadvantages of conventional liposomes is their rapid elimination from the bloodstream, and they arrive in organs and tissues of the reticuloendothelial system, such as the liver and spleen
[84][119]. The increases in liposome-circulating lifetimes promoted by PEG depend on the amount of grafted PEG and the molecular weight of the polymer. PEG longer chains typically increase the bloodstream residence time, reported as higher for PEGylated liposomes containing PEG 1900 and PEG 5000 compared to PEG 750 and PEG 120
[85][86][120,121]. Their molecular weight determines the conformation of PEG polymers on the surface of liposomes and PEG surface density in mushroom (low concentration) or brush (high concentration) regimes
[87][122]. PEG concentrations from 5% to 10% (molar ratio) result in improvements in the degree of liposome stealth, and higher PEG concentrations (brush regimen) input to liposomes lead to high resistance to phagocytosis and poor activation of the human complement system
[88][123].
PEG-enhanced liposome surfaces are associated with a cloaking effect, mimicking water-like structures, thus providing a steric barrier that prevents protein adsorption to liposome surfaces and their recognition by the mononuclear macrophage phagocytic system that would otherwise lead to rapid liposome clearance
[59][67][94,102]. On the other hand, repeated venous administrations of PEGylated liposomes in animals at certain intervals induce immune responses, resulting in the loss of long-circulating characteristics and accelerating the blood clearance (ABC) phenomenon
[89][90][124,125]. It has been suggested that anti-PEG IgM, produced by the spleen in response to a first dose, selectively binds to PEG chains in a second dose administered several days later and subsequently activates the complement system, one of the main opsonins, increasing the liver uptake of the following doses
[90][91][125,126]. The occurrence and magnitude of the ABC phenomenon are influenced by the dose and physicochemical properties of PEGylated liposomes and the time interval between liposome administration and the encapsulated drug
[92][127]. Many approaches have been tested to minimize the immunogenicity of the PEG moiety following repeated administrations. PEG lipids presenting a shorter alkyl chain can dissociate more quickly from the lipid bilayer, such as mPEG-DSPE and mPEG-CH, attenuating the ABC phenomenon
[93][94][128,129].