Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 3 by Jessie Wu.
Post-translational modifications (PTMs) are reversible or irreversible chemical reactions that occur in specific amino acids (aas) of some proteins after their translation. In fact, more than 140 different aa-based structures can constitute the proteins when considering the PTMs that could modify the 20 natural aas. Several factors, such as the location of the aa within the protein’s primary sequence, will determine whether PTMs occur as well as their type and frequency. PTMs are not encoded by the cellular genome and can significantly modify protein structure and modulate functions such as folding, degradation, signaling, localization, stability, enzymatic activity and protein-protein interactions. More than 400 different types of PTMs have been described in humans occurring in several cellular organelles (nucleus, cytoplasm, endoplasmic reticulum and Golgi apparatus). They are involved in a large number of cellular events and are considered a key mechanism for the regulation of the biological activity of proteins. The latest findings about the role of PTMs in the generation of autoimmunity is discussed. Meanwhile, the most relevant PTMs in rheumatic diseases that occur in synovial tissue are discussed.
- PTMs
- immune cells
- rheumatic diseases
- synthetic peptides
1. Post-Translational Modifications in the Generation of Autoimmunity
In the early 2000s, Doyle and Mamula clearly addressed the question of how post-translational modifications (PTMs) of proteins affect the processing of foreign and self-antigens and what their role is in the origin of autoimmune responses [1]. They shed light with explanatory examples on the role of PTMs in autoimmunity and detailed how protein modifications may alter protein biology, processing and presentation of autoantigens by the immune system as well as the mechanisms of the breakdown of tolerance by post-translationally modified proteins [2].
In the context of disease and inflammation, the generation of PTMs in human proteins is usually the consequence of a physiological response to cellular stress that can cause the release of cellular products such as reactive oxygen species (ROS) and enzymes that promote the modification of amino acid residues. The production of ROS at physiological levels contributes to the resolution of inflammation and the maintenance of homeostasis in tissues. However, when ROS release is excessive, these levels can increase to the point of overcoming the antioxidant capacity of natural defenses that are unable to eliminate excess ROS. In such conditions of oxidative stress, apoptosis processes also have a role in the generation of self-antigens since self-proteins are cleaved differently in apoptotic cells. Furthermore, these proteins are subject to several modifications such as phosphorylation, transglutamination, ubiquitination and citrullination. Therefore, cells undergoing apoptosis are considered rich sources of PTM-altered self-proteins [3].
The modified proteins can trigger the formation of new epitopes (neoepitopes) that are able to induce robust autoimmune responses altering the immune tolerance to self-proteins. The neoepitopes are different from the epitopes of self-proteins to which the immune system is already tolerant, becoming “foreign” to the organism and breaking tolerance of the immune system. In addition, it has been described that autoimmune responses originating from the post-translationally modified self-proteins diversify in an intra- and extra-molecular manner to other self-protein epitopes [4]. Thus, breaking immune tolerance to a post-translational modification (PTM) self-protein promotes “epitope spreading”, a mechanism where autoimmune response may be amplified involving other epitopes beyond the one which initiated the response. PTMs in self-proteins can profoundly affect the recognition of antigens by the antigen-presenting cells with the subsequent alteration of the specificity of B- and T-lymphocyte immunity. As described by Valesini et al. [5], small modifications in the structure can alter the immunogenicity of the proteins due to an enhanced protein unfolding and subsequent processing and exposure of the immunogenic epitopes [6].
As pointed out by Van Damme’s research group, autoimmunity research has been classically centered on the study of antigen presentation to T lymphocytes, the role of B lymphocytes and the antibody recognition of autoantigens. In addition to the adaptive immunity, these authors highlight the importance of the innate immune system in the generation of autoimmunity and propose that extracellular proteolysis, regulated by cytokines and proteases from innate immune cells, generates autoantigens that start autoimmune reactions [7]. Cytokine-regulated extracellular proteolysis and other PTMs of autoantigens can be executed by host enzymes, including complement molecules, matrix metalloproteinases and deiminases, demonstrating the prominent role that these molecules and innate immune cells play in autoimmune diseases [8].
In addition to the immune system, host genetics and environmental factors play also major roles in autoimmune pathogenesis and progression.
Regarding genetics, heritable genetic risk traits have been defined by Genome-Wide Association Studies (GWAS) in many autoimmune diseases. Recently, Caliskan et al. have reported a comprehensive catalog of GWAS fine mapping in autoimmune diseases with the aim of predicting the probability of mediating disease risk associations across genes in GWAS loci and identifying robust signals of causal disease biology [9].
Among environmental factors, microbes (bacteria and viruses) can induce PTMs that can contribute to autoantigenic neoepitope formation. Microorganisms can directly provide proteases or other enzymes that can modify specific amino acids in proteins or peptides via glycosylation or citrullination among other changes, which can contribute to the generation of autoantigens stimulating T cells. [10][11][12] One of the most-reported links between bacterial infection and autoimmune diseases through PTM is related to Porphyromonas gingivalis, the major pathogen of periodontal disease, with an increased prevalence in rheumatoid arthritis (RA) [13][14]. This prokaryote expresses the bacterial peptidylarginine deiminase (PAD) which generates citrullinated epitopes distinct from those generated by endogenous PADs, thus contributing to aggravation of RA [11]. More recently, in the context of the SARS-CoV-2 pandemic, it has been speculated that coronavirus infection might exacerbate the formation of PTMs and, in so doing, provoke the onset of type 1 diabetes mellitus [12]. Furthermore, Epstein-Barr virus (EBV) infection has been epidemiologically linked to multiple sclerosis [15]. High-affinity molecular mimicry between EBV nuclear antigen 1 (EBNA1) and glial cell adhesion molecule (GlialCAM) has been recently demonstrated, which is facilitated by post-translational phosphorylation of the central nervous system protein GlialCAM [16]. These latest findings highlight the role of viruses in autoimmune diseases and deserve further investigation to unravel the involvement of viruses in the processes that lead to inflammation.
2. Post-Translational Modifications in Inflammatory Immune-Mediated Rheumatic Diseases
Inflammatory rheumatic diseases are autoimmune and/or immune-mediated diseases caused by the immune system’s own attack on the joints, muscles, bones and organs. They include a variety of disorders involving synovial joints; among them RA and spondyloarthropathies (including psoriatic arthritis) are the most common, accounting for a large percentage of disability [17]. RA affects 0.5–1% of adults worldwide, with women being three times more susceptible than men. The chronic polyarthritis causes joint destruction and deformities, together with functional disability and reduced quality of life and life expectancy [18]. In RA there is a persistent activation of the immune system accompanied by inflammation, especially in the joints (synovitis), one of the main characteristics of said inflammation being ROS. This imbalance towards an excess of oxidant molecules (oxidative stress) is closely related to RA. Moreover, an interesting and enigmatic entity with a close, but not well understood, relationship with RA is Palindromic Rheumatism (PR), a form of relapsing/remitting arthritis that may evolve into RA. Although its frequency is significantly lower than that of RA it is clearly considered a pre-RA stage for most patients. PR autoimmunity plays a substantial role in this rheumatic disease, with the same characteristic autoantibody profile observed in RA, although with some differences in the immune response repertoire [19].
Comprehensively studied PTMs that occur in synovial tissue and at present considered more relevant in rheumatic diseases are citrullination, homocitrullination (carbamylation) and acetylation (Figure 1). Among them, citrullination is the best-studied PTM in rheumatology, and Anti-Citrullinated Protein/Peptide Antibodies (ACPAs) were included by the European League against Rheumatism (EULAR)/American College of Rheumatology (ACR) as classification criteria for RA in 2010 [20].
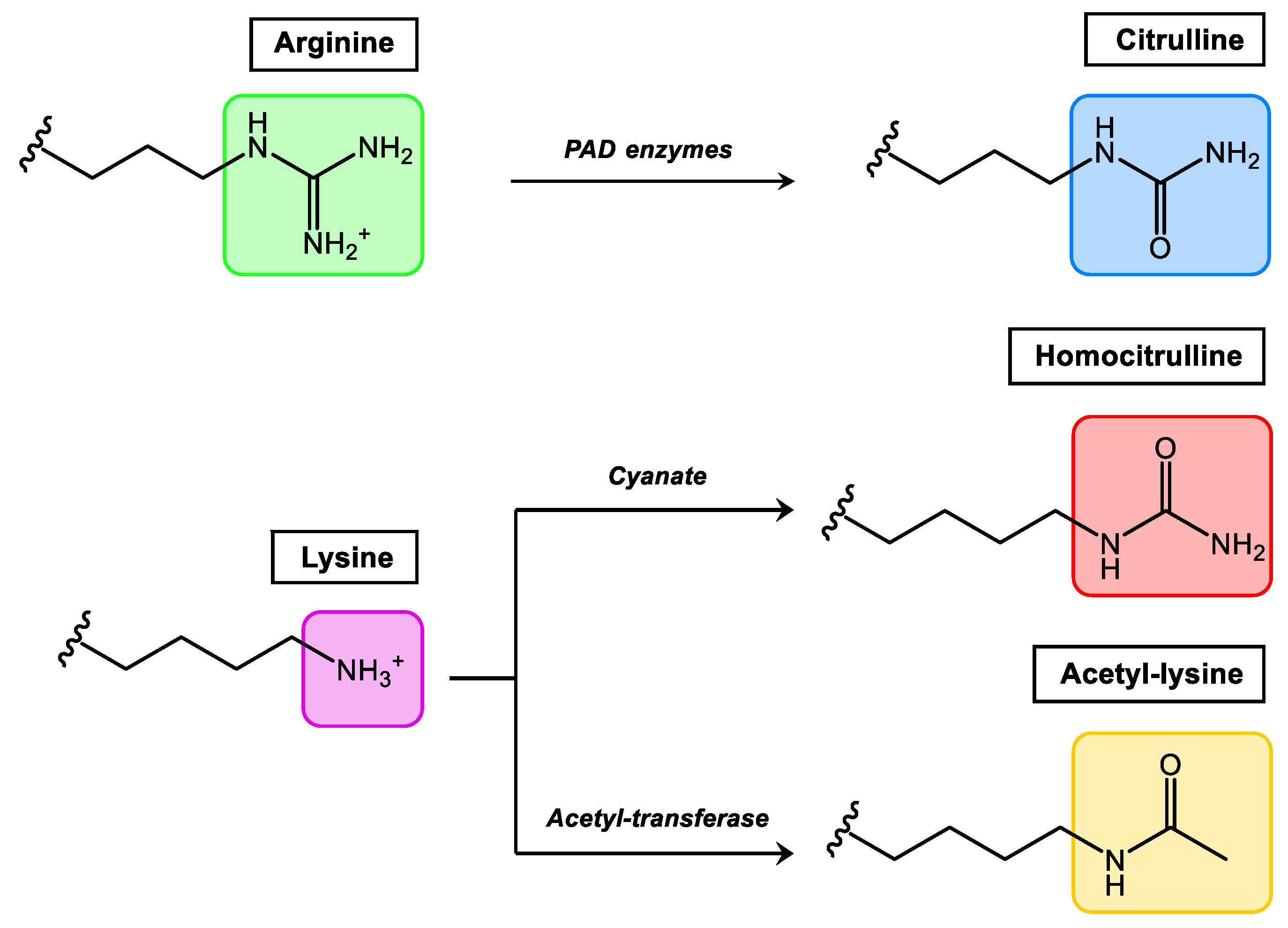
Figure 1. Relevant PTMs in inflammatory rheumatic diseases.
Citrullination, which consists of the conversion of arginine to the non-essential amino acid citrulline, is mediated by the enzyme peptidylarginine deiminase which is found in the inflamed synovium. Homocitrullination or carbamylation is a non-enzymatic PTM that involves the conversion of lysine residues to homocitrullinated residues after a reaction with cyanate. Acetylation is a reversible enzymatic process (balance between acetylases and deacetylases) where acetyl groups are added to free amines of lysine residues. In general terms, all autoantibodies found in RA belong to the Anti-Modified Protein/Peptide Antibodies (AMPAs) family.
Other autoantigens including malondialdehyde (MDA) and malondialdehyde-acetaldehyde (MAA) adduct containing proteins [21] or autoantibodies targeting the other four PTMs, such as chlorination, non-enzymatic glycation, nitration and homocysteinylation [22] have been recently studied in RA. Specifically, an increased lipid peroxidation is produced when tissues are exposed to oxidative stress, and this results in the formation of MDA which degrades into acetaldehyde and both are also able to react and form the more stable protein adduct MAA. However, despite the initially surprising described high sensitivity of anti-MAA antibodies for RA [21], these autoantibodies are not specific for RA, having been implicated in other diseases associated with increased oxidative stress and lipid peroxidation and detected in patients with other rheumatic conditions [23], cardiovascular diseases [24], liver damage [25], type II diabetes mellitus [26] or smoking-related diseases [27]. Recently, Rodriguez-Martinez et al. [22] reported only significant differences of anti-glycated collagen type II autoantibodies in RA patients, although the magnitude of the specific signal was very small, which led the authors to conclude that the four atypical antigens included in their analysis were not able to induce significant autoantibody response in RA patients, thus indicating that the repertoire of PTM autoantigens in RA is restricted.
Furthermore, N-glycosilation or O-glycosilation, the addition of glucides on an atom of nitrogen or oxygen of the lateral chain of the amino acid proteins, are mediated by hundreds of glycosyl-transferases and aberrant glycosylation of IgG has been implicated in RA pathogenesis [28][29].
ACPA target proteins include mainly endogenous autoantigens that are expressed in organs and tissues implicated in the immunopathology of RA [30][31][32]. Apart from filaggrin, one of the first citrullinated proteins identified as an ACPA target that is expressed in the epithelium [33], other highly expressed and citrullinated proteins have been described in inflamed synovium. These citrullinated antigens include structural components of the joints, such as type II collagen, α-enolase and fibronectin, proteins that form deposits in inflamed joints, mainly fibrinogen/fibrin and vimentin [34][35][36], as well as other antigens that have been identified in synovial fluid from RA patients (apolipoprotein E, β-actin and myeloid nuclear differentiation antigen or MNDA) [37]. In addition, intracellular proteins (immunoglobulin-binding protein, BIP) [38] or intranuclear proteins, [39][40][41] such as histones, chaperones and heterogeneous nuclear ribonucleoproteins (hnRNP), have also been described as targets of ACPAs in inflammatory conditions. These nuclear proteins become accessible to the immune system in the Neutrophil Extracellular Traps (NETs) [42] after being extruded by NETosis processes, which are another form of cell death different from apoptosis and necrosis.
References
- Doyle, H.A.; Mamula, M.J. Post-translational protein modifications in antigen recognition and autoimmunity. Trends Immunol. 2001, 22, 443–449.
- Doyle, H.A.; Mamula, M.J. Autoantigenesis: The evolution of protein modifications in autoimmune disease. Curr. Opin. Immunol. 2012, 24, 112–118.
- Utz, P.J.; Gensler, T.J.; Anderson, P. Death, autoantigen modifications, and tolerance. Arthritis Res. 2000, 2, 101–114.
- Yang, M.-L.; Sodre, F.M.C.; Mamula, M.J.; Overbergh, L. Citrullination and PAD Enzyme biology in type 1 diabetes—regulators of inflammation, autoimmunity, and pathology. Front. Immunol. 2021, 12, 678953.
- Valesini, G.; Gerardi, M.C.; Iannuccelli, C.; Pacucci, V.A.; Pendolin, M.; Shoenfeld, Y. Citrullination and autoimmunity. Autoimmun. Rev. 2015, 14, 490–497.
- Carrasco-Marin, E.; Paz-Miguel, J.; Lopez-Mato, P.; Alvarez-Dominguez, C.; Leyva-Cobian, F. Oxidation of defined antigens allows protein unfolding and increases both proteolytic processing and exposes peptide epitopes which are recognized by specific T cells. Immunology 1998, 95, 314–321.
- Opdenakker, G.; El-Asrar, A.A.; Van Damme, J. Remnant epitopes generating autoimmunity: From model to useful paradigm. Trends Immunol. 2020, 41, 367–378.
- Opdenakker, G.; Van Damme, J. Cytokine-regulated proteases in autoimmune diseases. Immunol. Today 1994, 15, 103–107.
- Caliskan, M.; Brown, C.D.; Maranville, J.C. A catalog of GWAS fine-mapping efforts in autoimmune disease. Am. J. Hum. Genet. 2021, 108, 549–563.
- Opdenakker, G.; Proost, P.; Van Damme, J. Microbiomic and posttranslational modifications as preludes to autoimmune diseases. Trends Mol. Med. 2016, 22, 746–757.
- Goulas, T.; Mizgalska, D.; Garcia-Ferrer, I.; Kantyka, T.; Guevara, T.; Szmigielski, B.; Sroka, A.; Millán, C.; Usón, I.; Veillard, F.; et al. Tructure and mechanism of a bacterial host-protein citrullinating virulence factor, Porphyromonas gingivalis peptidylarginine deiminase. Sci. Rep. 2015, 5, 11969.
- Chaplin, C.; Pozzilli, P. SARS-CoV-2 induced post-translational protein modifications: A trigger for developing autoimmune diabetes? Diabetes Metab. Res. Rev. 2022, 38, e3508.
- de Pablo, P.; Chapple, I.L.; Buckley, C.D.; Dietrich, T. Periodontitis in systemic rheumatic diseases. Nat. Rev. Rheumatol. 2009, 5, 218–224.
- Maresz, K.J.; Hellvard, A.; Sroka, A.; Adamowicz, K.; Bielecka, E.; Koziel, J.; Gawron, K.; Mizgalska, D.; Marcinska, K.A.; Benedyk, M.; et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PLoS Pathog. 2013, 9, e1003627.
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301.
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.-S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.-S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327.
- Sangha, O. Epidemiology of rheumatic diseases. Rheumatology 2000, 39, 3–12.
- Haro, I.; Sanmarti, R. Rheumatoid arthritis: Current advances in pathogenesis, diagnosis and therapy. Curr. Top. Med. Chem. 2013, 13, 697.
- Sanmartí, R.; Haro, I.; Cañete, J.D. Palindromic rheumatism: A unique and enigmatic entity with a complex relationship with rheumatoid arthritis. Expert. Rev. Clin. Immunol. 2021, 17, 375–384.
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League against rheumatism collaborative initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588.
- Thiele, G.M.; Duryee, M.J.; Anderson, D.R.; Klassen, L.W.; Mohring, S.M.; Young, K.A.; Benissan-Messan, D.; Sayles, H.; Dusad, A.; Hunter, C.D.; et al. Malondialdehyde-acetaldehyde adducts and anti-malondialdehyde-acetaldehyde antibodies in rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 645–655.
- Rodríguez-Martínez, L.; Regueiro, C.; Amhaz-Escanlar, S.; Pena, C.; Herbello-Hermelo, P.; Moreda-Piñeiro, A.; Rodriguez-Garcia, J.; Mera-Varela, A.; Pérez-Pampín, E.; González, A. Antibodies against 4 atypical post-translational protein modifications in patients with rheumatoid arthritis. Diagnostics 2022, 12, 352.
- Mikuls, T.R.; Duryee, M.J.; England, B.R.; Anderson, D.R.; Hearth-Holmes, M.; Su, K.; Michaud, K.; Payne, J.B.; Sayles, H.; Hunter, C.; et al. Malondialdehyde–acetaldehyde antibody concentrations in rheumatoid arthritis and other rheumatic conditions. Int. Immunopharmacol. 2018, 56, 113–118.
- Anderson, D.R.; Duryee, M.J.; Shurmur, S.W.; Um, J.Y.; Bussey, W.D.; Hunter, C.D.; Garvin, R.P.; Sayles, H.R.; Mikuls, T.R.; Klassen, L.W.; et al. Unique antibody responses to malondialdehyde-acetaldehyde (MAA)-protein adducts predict coronary artery disease. PLoS ONE 2014, 9, e107440.
- Rolla, R.; Vay, D.; Mottaran, E.; Parodi, M.; Traverso, N.; Aricó, S.; Sartori, M.; Bellomo, G.; Klassen, L.W.; Thiele, G.M.; et al. Detection of circulating antibodies against malondialdehyde-acetaldehyde adducts in patients with alcohol-induced liver disease. Hepatology 2000, 31, 878–884.
- Vehkala, L.; Ukkola, O.; Kesaniemi, Y.A.; Kahonen, M.; Nieminen, M.; Salomaa, V.; Jula, A.S.; Horkko, S. Plasma IgA antibody levels to malondialdehyde acetaldehyde-adducts are associated with inflammatory mediators, obesity and type 2 diabetes. Ann. Med. 2013, 45, 501–510.
- McCaskill, M.L.; Kharbanda, K.K.; Tuma, D.J.; Reynolds, J.D.; DeVasure, J.M.; Sisson, J.H.; Todd, A.; Wyatt, T.A. Hybrid malondialdehyde and acetaldehyde protein adducts form in the lungs of mice exposed to alcohol and cigarette smoke. Alcohol. Clin. Exp. Res. 2011, 35, 1106–1113.
- Parekh, R.B.; Dwek, R.A.; Sutton, B.J.; Fernandes, D.L.; Leung, A.; Stanworth, D.; Rademacher, T.W.; Mizuochi, T.; Taniguchi, T.; Matsuta, K.; et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 1985, 316, 452–457.
- Ercan, A.; Cui, J.; Chatterton, D.E.; Deane, K.D.; Hazen, M.M.; Brintnell, W.; O’Donnell, C.I.; Derber, L.A.; Weinblatt, M.E.; Shadick, N.A.; et al. Aberrant IgG galactosylation precedes disease onset, correlates with disease activity, and is prevalent in autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2239–2248.
- Fang, Q.; Ou, J.; Nandakumar, K.S. Autoantibodies as diagnostic markers and mediator of joint inflammation in Arthritis. Mediat. Inflamm. 2019, 2019, 6363086.
- Kwon, E.-J.; Ju, J.H. Impact of posttranslational modification in pathogenesis of rheumatoid arthritis: Focusing on citrullination, carbamylation, and acetylation. Int. J. Mol. Sci. 2021, 22, 10576.
- Tilvawala, R.; Nguyen, S.H.; Maurais, A.J.; Nemmara, V.V.; Mitesh Nagar, M.; Salinger, A.J.; Nagpal, S.; Weerapana, E.; Thompson, P.R. The Rheumatoid Arthritis-Associated Citrullinome. Cell Chem. Biol. 2018, 25, 691–704.
- Union, A.; Meheus, L.; Humbel, R.L.; Conrad, K.; Steiner, G.; Moereels, H.; Pottel, H.; Serre, G.; De Keyser, F. Identification of citrullinated rheumatoid arthritis-specific epitopes in natural filaggrin relevant for antifilaggrin autoantibody detection by line immunoassay. Arthritis Rheum. 2002, 46, 1185–1195.
- Ioan-Facsinay, A.; Willemze, A.; Robinson, D.B.; Peschken, C.A.; Markland, J.; van der Woude, D.; Elias, B.; Ménard, H.A.; Newkirk, M.; Fritzler, M.J.; et al. Marked differences in fine specificity and isotype usage of the anti-citrullinated protein antibody in health and disease. Arthritis Rheum. 2008, 58, 3000–3008.
- Snir, O.; Widhe, M.; Von Spee, C.; Lindberg, J.; Padyukov, L.; Lundberg, K.; Engström, A.; Venables, P.J.; Lundeberg, J.; Holmdahl, R.; et al. Multiple antibody reactivities to citrullinated antigens in sera from patients with rheumatoid arthritis: Association with HLA-DRB1 alleles. Ann. Rheum. Dis. 2008, 68, 736–743.
- Van Beers, J.J.; Willemze, A.; Stammen-Vogelzangs, J.; Drijfhout, J.W.; Toes, R.E.; Pruijn, G.J.M. Anti-citrullinated fibronectin antibodies in rheumatoid arthritis are associated with human leukocyte antigen-DRB1 shared epitope alleles. Arthritis Res. Ther. 2012, 14, R35.
- Van Beers, J.J.; Schwarte, C.M.; Stammen-Vogelzangs, J.; Oosterink, E.; Bozic, B.; Pruijn, G.J.M. The rheumatoid arthritis synovial fluid citrullinome reveals novel citrullinated epitopes in apolipoprotein E, myeloid nuclear differentiation antigen, and β-actin. Arthr. Rheum. 2013, 65, 69–80.
- Shoda, H.; Fujio, K.; Shibuya, M.; Okamura, T.; Sumitomo, S.; Okamoto, A.; Sawada, T.; Yamamoto, K. Detection of autoantibodies to citrullinated BiP in rheumatoid arthritis patients and pro-inflammatory role of citrullinated BiP in collagen-induced arthritis. Arthritis Res. Ther. 2011, 13, R191.
- Meng, X.; Ezzati, P.; Smolik, I.; Bernstein, C.N.; Hitchon, C.A.; El-Gabalawy, H.S. Characterization of autoantigens targeted by anti-citrullinated protein antibodies in vivo: Prominent role for epitopes derived from Histone 4 proteins. PLoS ONE 2016, 11, e0165501.
- Goëb, V.; Thomas-L’Otellier, M.; Daveau, R.; Charlionet, R.; Fardellone, P.; Le Loët, X.; Tron, F.; Gilbert, D.; Vittecoq, O. Candidate autoantigens identified by mass spectrometry in early rheumatoid arthritis are chaperones and citrullinated glycolytic enzymes. Arthritis Res. Ther. 2009, 11, R38.
- Konig, M.F.; Giles, J.T.; Nigrovic, P.A.; Andrade, F. Antibodies to native and citrullinated RA33 (hnRNP A2/B1) challenge citrullination as the inciting principle underlying loss of tolerance in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 2022–2028.
- Wright, H.; Moots, R.; Edwards, S. The multifactorial role of neutrophils in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 593–601.
More