Abstract: Glioblastoma multiforme is the most malignant and aggressive type of brain neoplasm, with a mean life expectancy of less 15 months after diagnosis, despite a diversity of treatments, including surgery, radiation, chemotherapy, and immunotherapy. The resistance of GBM to various therapies is due to a highly mutated genome; these genetic changes induce a de-regulation of several signaling pathways and result in higher cell proliferation rates, angiogenesis, invasion, and a marked resistance to apoptosis; this latter trait is a hallmark of highly invasive tumor cells, such as glioma cells. Due to a defective apoptosis in gliomas, induced autophagic death can be an alternative to remove tumor cells. Paradoxically, however, autophagy in cancer can promote either a cell death or survival. Modulating the autophagic pathway as a death mechanism for cancer cells has prompted the use of both inhibitors and autophagy inducers. The autophagic process, either as a cancer suppressing or inducing mechanism in high-grade gliomas is discussed in this section.
- glioblastoma
- autophagic pathway
- autophagic death
1. Definition
Autophagy is a catabolic process that facilitates the recycling of cellular constituents in response to stressing conditions, such as nutrient deprivation or infection; thus, promoting the recovery of cellular balance. It has been classified into macroautophagy, microautophagy, and chaperone-mediated autophagy [1]. Macroautophagy involves training an autophagosome, a vesicle that will surround the proteins or organelles to be recycled and fuse with the lysosome to degrade the cargo; microautophagy involves invagination of the lysoome, with the target proteins or organelles inside it. In chaperone-mediated autophagy, the heat-shock cognate 70-kDa (HSC70) protein will recognize the KFERQ-motif in the target proteins and facilitate their translocation into the lysosome through the lysosomal-associated membrane protein 2A (LAMP2A) receptor.
2. Introduction
Glioma is the most frequent and aggressive type of brain neoplasm, being anaplastic astrocytoma (AA) and glioblastoma multiforme (GBM), its most malignant forms. The resistance of GBM to various therapies is due to a highly mutated genome; these genetic changes induce a de-regulation of several signaling pathways and result in higher cell proliferation rates, angiogenesis, invasion, and a marked resistance to apoptosis; this latter trait is a hallmark of highly invasive tumor cells, such as glioma cells. Due to a defective apoptosis in gliomas, induced autophagic death can be an alternative to remove tumor cells.
3. Pathways and Molecular Mechanisms of Autophagy
Autophagy is a catabolic process that facilitates the recycling of cellular constituents in response to stressing conditions, such as nutrient deprivation or infection; thus, promoting the recovery of cellular balance. It has been classified into macroautophagy, microautophagy, and chaperone-mediated autophagy. This entry will focus on macroautophagy, referred simply as autophagy from this point on. Autophagy comprises the following stages: induction, nucleation, elongation, and completion; fusion to lysosome, demotion, and recycling [21]. The process involves a large complex of proteins that interact among them in response to inhibitory or activating conditions (Figure 1).
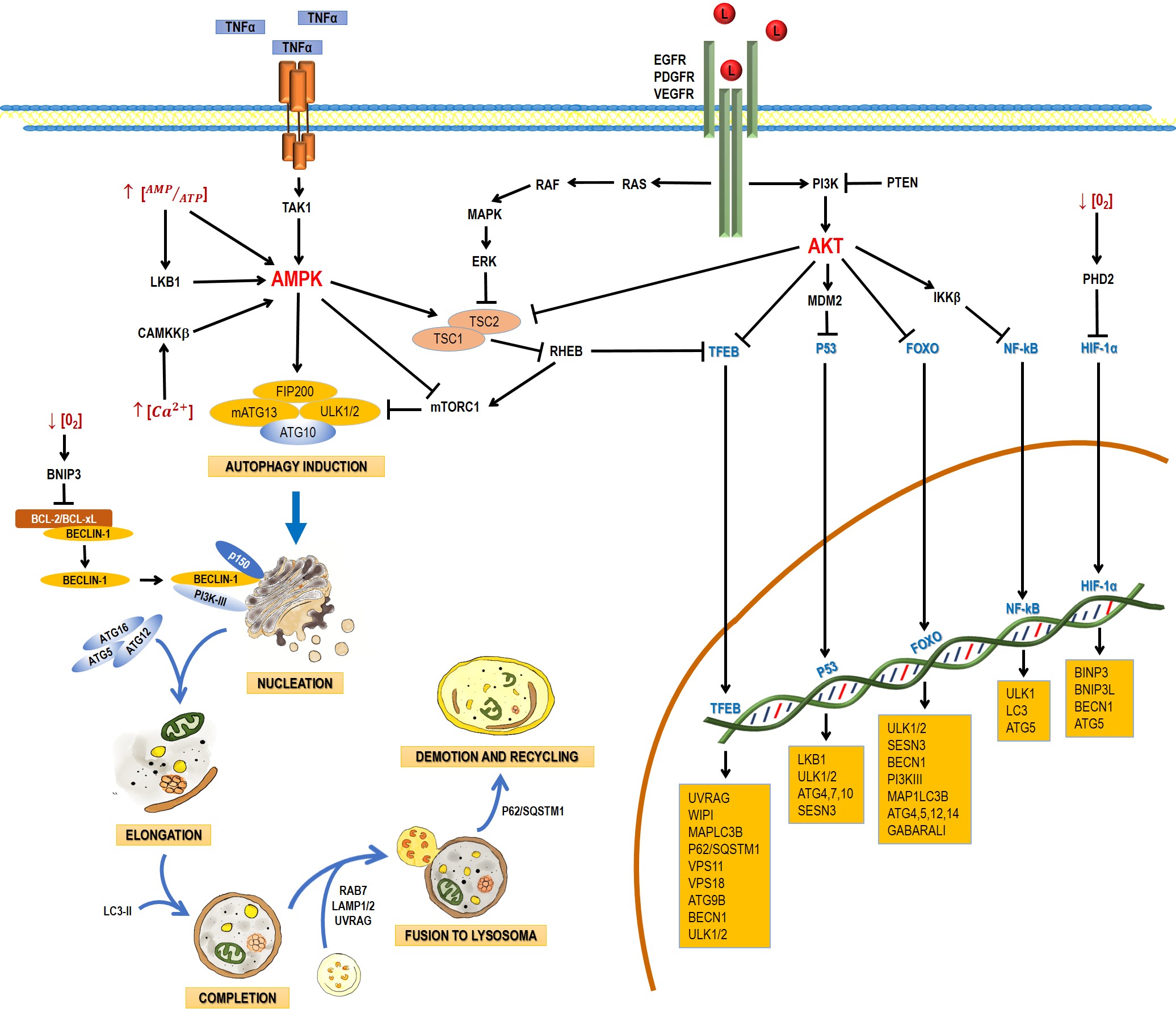
Figure 1. Schematic process of the autophagic pathway. Autophagy consists of the following stages: induction, phagophore nucleation, elongation, and completion; fusion to lysosome, degradation, and recycling. Receptor tyrosine kinases (EGF (epidermal growth factor receptor), PDGF (platelet-derived growth factor receptor), and VEGF (vascular endothelial growth factor receptor)) stimulate the RAS/RAS/ERK and PI3K/AKT signaling pathways, which traduce signals to activate transcription factors, such as transcription factor EB (TFEB), FOXO, P53, and NF-кB, which regulate the expression of genes that are key for autophagy induction. Then, the activation of AKT inhibits autophagic death by activating the mTORC1 complex, which contains mTOR. During nutrient deprivation, AMPK stimulates autophagy by phosphorylation of ULK1 and suppression of mTORC1, which inhibits autophagy by inactivating ULK1. Autophagy can also be induced by hypoxic conditions through HIF activation. Autophagosome biogenesis starts with the formation of an initiation membrane, derived from the endoplasmic reticulum (ER) or other cellular membrane sources. Vesicle nucleation requires the activity of the PI3K-III/Beclin-1 complex. The completion and expansion of the autophagosome requires the Atg5/Atg12/Atg16 and LC3-PE proteins. Mature autophagosome fuses with a lysosome to form the autophagosome, which requires the action of proteins, such as Rab7, UVRAG, and Lamp 1, 2; finally, p62/SQSTM1 is required for the degradation and recycling of the cellular components. Black arrows (↓) indicate activation, and black truncated arrows (⊥) indicate inhibition. Blue arrows (↓) indicate the process of the autophagic pathway.
3.1. Induction
In yeasts, autophagy induction requires the inhibition of the mammalian target of rapamycin complex 1 (mTORC1) and the activation of the canonical autophagy pathway, involving the Atg genes. In nutrient-rich media, mTOR activation leads to the hyperphosphorylation of Atg13 (mammalian homologue: ATG13), preventing thus its association to Atg1 (mammalian homologue: unc-51-like kinase 1 and 2 (ULK1 and ULK2)) and increasing its interaction with Atg11. During nutrient deprivation or treatment with rapamycin (mTORC1 inhibitor), Atg13 is hypophosphorylated, leading to the interaction between Atg1 and Atg13, triggering autophagy [32].
3.2. Nucleation
Several studies have suggested that nucleation takes place in the endoplasmic reticle in mammal cells. Autophagosome formation requires a vesicle to be formed through the Atg6 complex (mammalian homologue: Beclin-1), Vps34 (mammalian homologue: PI3K-III), Atg14, and Vps15 (mammalian homologue: p150) and the complex responsible for vesicle nucleation, which includes Atg1, Atg11, Atg13, and Atg17. The process of autophagosome formation is not well understood and new elements are integrated progressively. For example, the multispanning membrane protein Atg9 has been found to be essential for the autophagosome formation in yeast. Apparently, some Atg9 vesicles derived from the Golgi apparatus are able to assemble into PAS and become part of the autophagosomal outer membrane, with the possibility to be recycled later. In mammals, mAtg9 is highly conserved but, unlike yeast proteins, mAtg9 is
only temporary part of autophagosomes and does not integrate to autophagosomal membranes [43].
3.3. Elongation and Completion
Autophagosome elongation and completion occurs through two different conjugation pathways: Atg8-Atg4 and Atg12-Atg5-Atg16. Both pathways regulate lipid (phosphatidylethanolamine, PE) conjugation to Atg8 (mammalian homologue: MAP-LC3). In the Atg12-Atg5-Atg16 pathway, Atg12 is activated by Atg7, which allows Atg10 to be transferred [54] and bind Atg5 [6]. The Atg12-Atg5 complex binds Atg16. This trimer oligomerizes and locates itself on the autophagosome outer surface. The Atg12/Atg5/Atg16 complex mediates the binding of LC3-PE to the autophagosome membrane [65].
3.4. Fusion with Lysosomes and Degradation
After maturation, autophagosomes are fused to lysosomes to form the autophagolysosome, which requires the action of several proteins, such as Rab7 and Vti1p in mammalian cells [76][87][98].
In mammals, the fusion of autophagosomes to lysosomes depends on the soluble NSF attachment protein (SNARE) receptor, being syntaxin 17 (Stx17) a key protein in this step. After its recruitment to autophagosomes, Stx17 forms a trans-SNARE complex to allow membrane fusion. The recruitment of Stx17 to the autophagosomes is crucial. Additionally, a direct interaction of Stx17 with the small guanosine-triphosphatase named as immunity-related GTPase M (IRGM) and of both with Atg8 proteins seems to be required for autophagosome assembly; this complex, named as autophagosome recognition particle (ARP), is critical in this early step of autophagolysosome formation [109].
UVRAG plays another role in later autophagy stages: it regulates autophagosome maturation independently of Beclin-1. UVRAG facilitates the recruitment of the C vacuolar protein (C-VPS) to the autophagosome. The ensuing autophagosome-lysosome fusion leads to a rupture of the inner membrane and a degradation of the cytosolic content by lysosomal hydrolases. The interaction of UVRAG with the C-VPS complex promotes the activity of a Rab7-GTPase; in conjunction with proteins, such as LAMP-1 and LAMP-2, this results in the fusion of the autophagosome and lysosome [110].
4. Regulation of Autophagic Process
4.1. Autophagy Regulation for AMPK and mTOR
The main autophagy regulators are the 5ʹ-AMP-activated protein kinase (AMPK) and the serine/threonine protein-kinase (mTOR) (Figure 1). mTOR has been reported to be inactivated by a lack of nutrients, growth factor deprivation, rapamycin administration, or stress. These factors inhibit protein synthesis and activate autophagy to obtain energy [121]. The TORC1 complex is mainly regulated by AMPK (inhibitor), ERK, and AKT (activators) (Figure 1). The presence of growth factors (EGF and PDGF) leads to the formation of PIP3 via PI3K, with the ensuing activation of AKT. Growth factors also lead to the activation of ERK via RAS/MEK/ERK. ERK and AKT induce phosphorylation of TSC2, preventing the TSC2/TSC1 complex to be formed and the activation of TORC1. TORC1 blocks the initiation of autophagy through the phosphorylation (inactivation) of Atg13, ULK, AMBRA, and Atg-14L, inhibiting the complex Atg13-ULK1 and the activity of ULK1 and vps34.
4.2. Regulation of autophagy at a nuclear level by transcription factors
4.2.1. P53
Autophagy can also be regulated by transcription factors. The tumor-suppressing gene TP53 is activated by conditions like DNA damage or oxidative stress and P53 seems to promote the autophagic processes by enhancing the expression of genes involved in autophagy induction (LKB1, ULK1/2) and autophagosome maturation (ATG4, ATG7 and ATG10). It also activates the expression of Sestrin, a protein that activates AMPK [132]. p53 also increases the expression of the damage-regulated autophagy modulator (DRAM), which activates autophagy and apoptosis in a concerted manner [143].
4.2.2. TFEB
The transcription factor EB (TFEB) is linked primarily to lysosome biogenesis, but it also modulates the activation of autophagy in starvation [154]. mTOR, ERK2, and GSK3B are the main kinases that phosphorylate and sequester TFEB in the cytoplasm [165]; but when TFEB is dephosphorylated, it is translocated to the nucleus and controls processes like autophagosome formation and autophagosome-lysosome fusion.
4.2.3. FoxO
The Forkhead box class O (FoxO) family of transcription factors is another autophagy regulator. FoxO proteins are usually located in the cytoplasm and translocate to the nucleus to induce the expression of several genes, including those that regulate autophagy induction (ULK1, ULK2, SESN3), nucleation (BECN1, ATG14, PI3KIII), elongation (MAP1LC3B, ATG4, ATG5, ATG12, GABARALI), and autophagosome-lysosome fusion (Rab7 and TFEB, another transcription factor) [176].
4.2.4. HIF-1a
Autophagy can also be induced by hypoxia, and HIF-1a is key in this process. HIF-1a induces mitochondrial autophagy (mitophagy) linked with the expression of BINP3, BNIP3L, Beclin-1, and Atg5 [187]. BINP3 also competes with Beclin-1, impairing the interaction of Beclin-1 with Bcl2 and releasing Beclin1 to activate autophagy [198].
5. Autophagy in Glioma
Autophagy has been proposed as an GBM suppressing by removing damaged proteins and organelles, protecting cells from genomic instability, and metabolic alterations, inhibiting the initial phase of the carcinogenic process[2019]. On the other hand, glioma cells promote autophagy under adverse circumstances to sustain their survival, evading the physiological response to cancer and therapy, allowing tumor progression [210][221] (Figure 2). Autophagy could impact on the prognosis of glioma, either positively or negatively.
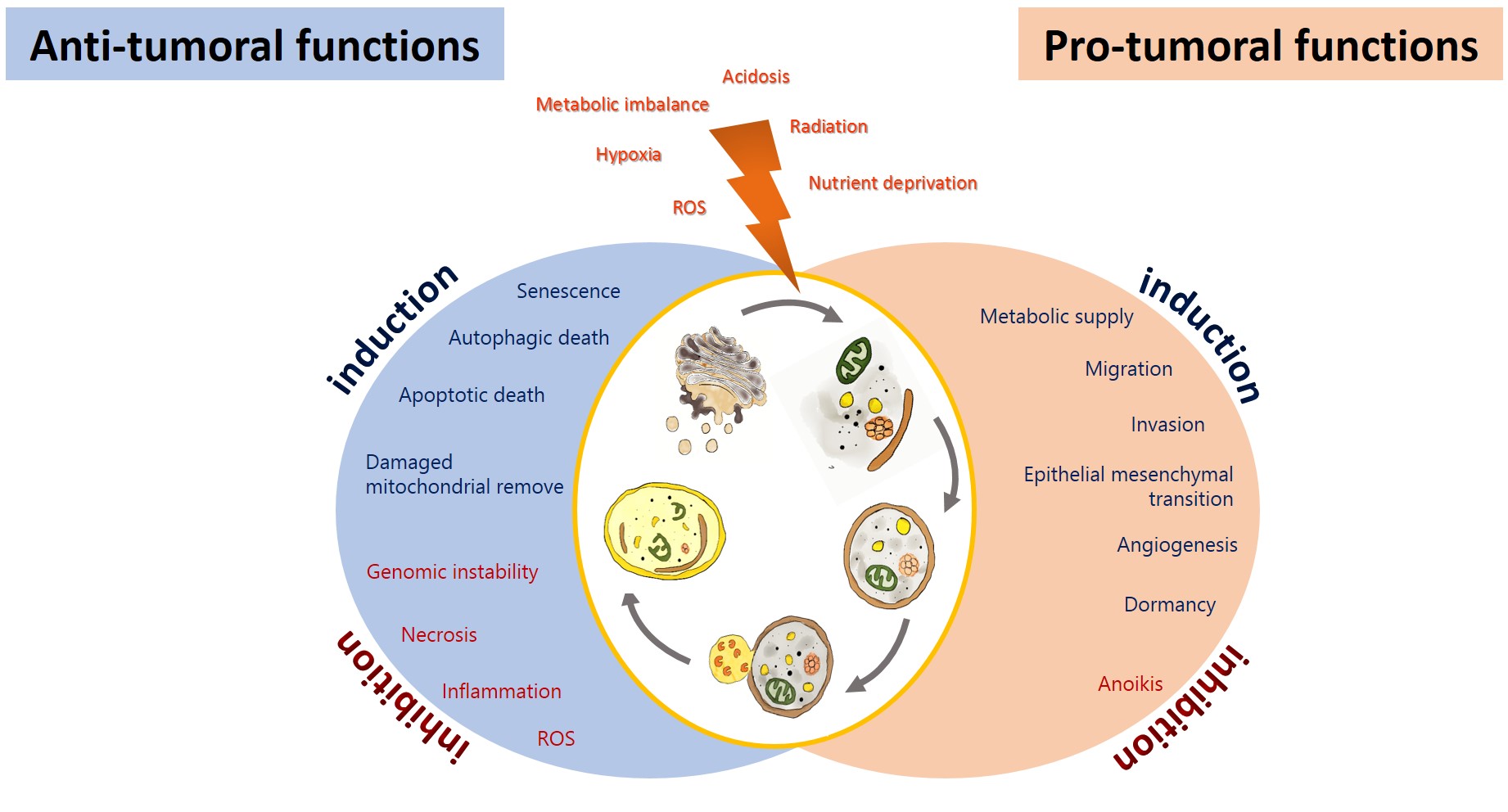
Figure 2. Anti-tumoral and pro-tumoral functions of autophagy. As an anti-tumoral process, autophagy prevents the accumulation of p62 aggregates, damaged proteins, and mitochondria, which may lead to ROS generation and the activation of oncogenic signaling pathways to stimulate necrosis, inflammation, and genomic instability, which lead to malignant transformation, tumor cell proliferation, migration, and invasion. Excessive autophagy can lead to type-II programmed cell death, which can induce apoptosis and activate senescence. On the other hand, autophagy sustains tumor growth and survival under adverse conditions (hypoxia and metabolic, osmotic, and oxidative stress). Uncontrolled proliferation leads to degrading misfolded or unnecessary proteins and organelles to mobilize amino acids, lipids, and carbohydrates, promoting cell survival. It also increases oncogenic signals that favor metabolism (glycolytic functions), angiogénesis, migration, and invasión, additionally, it inhibits anoikis and dormancy.
5.1. Autophagy as a Tumor suppressor in Glioma
Autophagy has been shown to act as a tumor suppressor in glioma, since the progression of astrocytic tumors is associated with a decrease in autophagic capacity [223]. Shukla et al. reported that ULK1 and ULK2 mRNA and protein levels are significantly decreased in glioblastoma patients with respect to normal brain samples, promoting astrocytic transformation by impairing autophagy [243]. Additionally, a lower expression or deletion of important genes for autophagosome initiation and elongation, such as FIP200, Beclin-1, UVRAG, Bif1 Atg4c, and Atg5 has been reported in GBM [243]. Lower levels of Beclin-1 transcript and protein were reported in glioblastoma [254]. It is noteworthy that the increased levels of the proteins LC3 and Beclin-1 were associated with an improved survival in GBM patients with poor performance scores [265].
Autophagy may exert its tumor-suppressing function in various ways (Figure 2). Autophagy prevents the accumulation of damaged and mitochondria, protecting the cell from oxidative stress and the subsequent genomic instability, necrosis, and inflammation that lead to tumor migration and invasion [276]. If autophagy is excessive, this can lead to type-II programmed cell death, which can induce apoptosis. In addition to activate senescence [287]. Cellular senescence is a steady status of proliferative detention that limits malignant transformation. It has been reported that TMZ induced autophagy followed by senescence in glioma cells [298]. Furthermore, autophagy may suppress tumorigenesis by removing p62-tagged aggregates. p62 accumulation causes DNA, protein, and mitochondrial damage and the generation of ROS [3029]. An overexpression of p62 has been reported in GBM patients, correlated with a poorer prognosis [276]. Another mechanism by which autophagy can prevent the carcinogenic process involves inhibiting or reverting EMT. EMT and its reverse process (MET) are both essential for cancer migration, invasion, chemo- and radioresistance [310]. Autophagy induced by amino acids and rapamycin in GL15 and U87 glioma cells was reported to revert EMT, reducing tumor cell migration and invasion by inhibiting the synthesis of SNAIL and increasing the expression of N-cadherin and R-cadherin [321].
5.2. Autophagy as a Tumoral Promotor in Glioma
It has been demonstrated that the induction of autophagy by stress in tumor cells can result in resistance to the treatment, with the consequent tumor recurrence and progression [332]. Tamrakar et al. demonstrated that a marked increase in the expression of p62, LC3, and Beclin-1 was related with radiation therapy in glioblastoma biopsy samples, whereas LC3 and p62 expression was associated with a poorer overall survival, and LC3 was associated with the methylation in the promoter of O6-methylyguanine-DNA methyltransferase (MGMT) and telomerase reverse transcriptase (TERT) [343]. On the other hand, a higher expression of LC3/Beclin-1 correlated with a shorter progression-free survival in low- and high-grade glioma patients [354]. Wen et al. found that a higher ATG4C transcript expression in patients with high-grade glioma correlated with a reduced overall survival (OS). ATG4C knockdown in T98G glioma cells inhibited autophagy, enhancing the cell cycle arrest and promoting apoptosis through the production of ROS, the expression of p21, p53, and Bax, and decreased levels of Bcl-2 [365]. Several mechanisms have been proposed by which autophagy favors tumorigenesis (Figure 2): Autophagy favors the survival of tumor cells in hypoxic areas (insufficient vascularization, limited supply of oxygen and nutrients) in solid tumors, such as glioblastoma, by degrading proteins, membranes, lipid droplets, and organelles to produce amino acids, fatty acids, and metabolic substrates that will allow tumor proliferation and survival [376]. Hypoxia (~3–0.1% oxygen) induces the activation of the hypoxia-inducible factor 1-alpha (HIF-1α), which promotes autophagy through the transcription regulation of autophagic genes, such as that coding for the Bcl-2/E1B 19-kDa-interacting protein (BNIP3), as well as BNIP3L, BECN1, and ATG5 [376]. BNIP3/BNIP3L induces autophagy through the release of Beclin-1 from the Bcl-2/Beclin-1 or Bcl-xL/Beclin-1 complexes[187]. The tumor microenvironment promotes the growth of cancer cells via autophagy. Tumor cells lead to an increase in ROS generation, inducing autophagy in fibroblasts; in turn, this promotes glycolysis in stroma cells, increasing the levels of pyruvate, lactate, acetoacetate, and 3-hydroxy- butyrate, which cover the nutritional and energetic requirements of tumor cells, inhibiting apoptosis, and increasing the growth and metastasis of tumor cells [187]. Furthermore, oxidative stress in tumor cells induces the activation of pro-autophagy factors, such as LC3, BNIP3L, ATG16L, BNIP3, NF-κB, and HIF-1α, which promote the degradation of caveolin-1 (Cav-1), leading to autophagy activation. Cav-1 acts as a spontaneous suppressor of autophagy by binding (inactivating) the autophagic proteins ATG5, ATG12, the ATG12-ATG5 complex, and LC3B, as well as by modulating the expression of ATG16L, ATG5, ATG12, and LC3 [387]. Autophagy has also been reported to facilitate the dissemination of tumor cells, favoring invasion and metastasis, by inhibiting anoikis. Anoikis is a form of programmed cellular death linked to detachment from the extracellular matrix (ECM) [398]. Under stress conditions, solid tumors, such as glioblastoma, exhibit resistance to cell death by inducing autophagy in cells detached from the primary tumor through PERK and the subsequent activation of the activating transcription factor 4 (ATF4), which induces the expression of autophagic genes, including ATG5, ATG7, and ULK, and of the antioxidant enzyme hemoxygenase-1, preventing anoikis and favoring the survival and migration of tumor cells [4039]. On the other hand, autophagy under conditions of metabolic stress (lack of growth factors, nutrients, and oxygen) can extend the survival of apoptosis-deficient cancer cells, promoting a period of dormancy or quiescence and prompting cell proliferation once the stressing stimulus is over [410]. Cellular dormancy is characterized by a reversible arrest in the growth of single cells or cell groups as a response to stressing agents, such as hypoxia or cytotoxic drugs [421]; dormant cells can remain hidden and asymptomatic for a long time. Dormancy induction mediated by HIF-1α segregated by stem-like tumor cells in the peri-necrotic niche favors tumoral recurrence, decreasing the survival of GBM patients that received the standard therapy (surgery followed by radiation and chemotherapy [TMZ]) [432].
6. Conclusions
Glioblastoma multiforme (GBM) is one of the most difficult neoplasms to treat due to its heterogeneity, tumor microenvironment, and invasiveness, which allows neoplastic cells to infiltrate the brain parenchyma, preventing a total resection of the tumor. It has been suggested that pharmacologic resistance in GBM is due to a high concentration of antiapoptotic proteins (Bcl-2, Bcl-xL and Mcl-1) and a low expression of proapoptotic proteins (BAX, Bak, pBad, pBim, and Survivin), favoring the switching-on of oncogenes and genetic instability that promote tumor survival and resistance to radiotherapy, chemotherapy, and immunotherapy. Cell death by autophagy has emerged as a promising strategy to remove neoplastic cells. Evidence indicates that autophagy mediates oncosuppressive and oncogenic effects in glioma. However, it is not easy to discern to what extent autophagy results in a positive or negative balance for cell survival, and therefore it is crucial to interfere with autophagy in a specific manner, adapted to the molecular, cellular, and epigenetic context, to the heterogeneity of tumor cells and microenvironment, and to the location of high-grade gliomas. Future translational studies on the interrelation and regulation of autophagy in tumor cells, glioma stem cells, and tumor microenvironment in a genic, epigenetic, and metabolic level will allow us to manipulate the complex processes of autophagy to devise therapies (more selective inhibitors of autophagic processes) for high-grade gliomas.
References
- Autophagy At The Crossroads Of Catabolism And Anabolism . Pubmed. Retrieved 2020-7-26Histological Characterization of the Tumorigenic "Peri-Necrotic Niche" Harboring Quiescent Stem-Like Tumor Cells in Glioblastoma . pubmed. Retrieved 2020-7-27
- Tor-mediated induction of autophagy via an Apg1 protein kinase complex . PUBMED. Retrieved 2020-7-27
- Tor-mediated induction of autophagy via an Apg1 protein kinase complex . pubmed. Retrieved 2020-7-26
- Atg9 vesicles are an important membrane source during early steps of autophagosome formation . pubmed. Retrieved 2020-7-26
- Mammalian Autophagy: Core Molecular Machinery And Signaling Regulation. . pubmed. Retrieved 2020-7-26
- Eaten Alive: A History Of Macroautophagy . pubmed. Retrieved 2020-7-27
- Exploring The Role Of Autophagy-Related Gene 5 (Atg5) Yields Important Insights Into Autophagy . pubmed. Retrieved 2020-7-27
- Rab7 Is Required For The Normal Progression Of The Autophagic Pathway In Mammalian Cells . pubmed. Retrieved 2020-7-27
- Role for Rab7 in maturation of late autophagic vacuoles . pubmed. Retrieved 2020-7-27
- Mechanism Of Stx17 Recruitment To Autophagosomes Via Irgm And Mammalian Atg8 Proteins. . pubmed. Retrieved 2020-7-27
- Beclin1-Binding Uvrag Targets The Class C Vps Complex To Coordinate Autophagosome Maturation And Endocytic Trafficking . pubmed. Retrieved 2020-7-27
- Mammalian Target Of Rapamycin And S6 Kinase 1 Positively Regulate 6-Thioguanine-Induced Autophagy . pubmed. Retrieved 2020-7-27
- Overexpression Of Dram Enhances P53-Dependent Apoptosis . pubmed. Retrieved 2020-7-27
- Transcriptional Regulation Of Autophagy: Mechanisms And Diseases. . pubmed. Retrieved 2020-7-27
- MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB . pubmed. Retrieved 2020-7-27
- TFEB links autophagy to lysosomal biogenesis . pubmed. Retrieved 2020-7-27
- The FoxO-Autophagy Axis in Health and Disease . pubmed. Retrieved 2020-7-27
- Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains . pubmed. Retrieved 2020-7-27
- Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia . pubmed. Retrieved 2020-7-27
- Autophagy in human health and disease . pubmed. Retrieved 2020-7-27
- Autophagy and Tumor Metabolism . pubmed. Retrieved 2020-7-27
- Autophagy-mediated tumor promotion . pubmed. Retrieved 2020-7-27
- Reduced expression of LC3B-II and Beclin 1 in glioblastoma multiforme indicates a down-regulated autophagic capacity that relates to the progression of astrocytic tumors . pubmed. Retrieved 2020-7-27
- Methylation silencing of ULK2, an autophagy gene, is essential for astrocyte transformation and tumor growth . pubmed. Retrieved 2020-7-27
- Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours . pubmed. Retrieved 2020-7-27
- Monitoring autophagy in glioblastoma with antibody against isoform B of human microtubule-associated protein 1 light chain 3 . pubmed. Retrieved 2020-7-27
- The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells . pubmed. Retrieved 2020-7-27
- Autophagy mediates the mitotic senescence transition . pubmed. Retrieved 2020-7-27
- Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage . pubmed. Retrieved 2020-7-27
- Autophagy suppresses tumorigenesis through elimination of p62 . pubmed. Retrieved 2020-7-27
- Regulatory networks defining EMT during cancer initiation and progression . pubmed. Retrieved 2020-7-27
- Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells . pubmed. Retrieved 2020-7-27
- Autophagy in brain tumors: a new target for therapeutic intervention . pubmed. Retrieved 2020-7-27
- Clinicopathological Significance of Autophagy-related Proteins and its Association With Genetic Alterations in Gliomas . pubmed. Retrieved 2020-7-27
- High LC3/Beclin Expression Correlates with Poor Survival in Glioma: a Definitive Role for Autophagy as Evidenced by In Vitro Autophagic Flux . pubmed. Retrieved 2020-7-27
- Knockdown ATG4C inhibits gliomas progression and promotes temozolomide chemosensitivity by suppressing autophagic flux . pubmed. Retrieved 2020-7-27
- Hypoxia-induced autophagy: cell death or cell survival? . pubmed. Retrieved 2020-7-27
- Interaction of caveolin-1 with ATG12-ATG5 system suppresses autophagy in lung epithelial cells . pubmed. Retrieved 2020-7-27
- Detachment-induced autophagy during anoikis and lumen formation in epithelial acini . pubmed. Retrieved 2020-7-27
- ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis . pubmed. Retrieved 2020-7-27
- Growth factor regulation of autophagy and cell survival in the absence of apoptosis . pubmed. Retrieved 2020-7-27
- Tumour cell dormancy as a contributor to the reduced survival of GBM patients who received standard therapy . pubmed. Retrieved 2020-7-27
- Histological Characterization of the Tumorigenic "Peri-Necrotic Niche" Harboring Quiescent Stem-Like Tumor Cells in Glioblastoma . pubmed. Retrieved 2020-7-27