Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Karolina Urbańska.
Hyperinsulinaemic hypoglycaemia (HH) is the most common cause of persistent hypoglycaemia in infants and children with incidence estimated at 1 per 50,000 live births. Congenital hyperinsulinism (CHI) is symptomatic mostly in early infancy and the neonatal period. Symptoms range from ones that are unspecific, such as poor feeding, lethargy, irritability, apnoea and hypothermia, to more serious symptoms, such as seizures and coma. During clinical examination, newborns present cardiomyopathy and hepatomegaly. The diagnosis of CHI is based on plasma glucose levels <54 mg/dL with detectable serum insulin and C-peptide, accompanied by suppressed or low serum ketone bodies and free fatty acids.
- congenital hyperinsulinaemic hypoglycaemia
- congenital hyperinsulinism
- ABCC8
- neonatal hypoglycaemia
1. Introduction
Hyperinsulinaemic hypoglycaemia (HH) is the most common cause of persistent hypoglycaemia in infants and children [1]. It is characterised by dysregulated insulin secretion from pancreatic β-cells despite low glucose levels [2]. It can be either congenital or acquired. Congenital hyperinsulinism mutations in genes related with the regulation of insulin secretion have been described. These genes are ABCC8, KCNJ11, GLUD1, GCK, HADH, UCP2, SLC16A1, HNF4A, HNF1A, HK1, PGM1 and PMM2 (Table 1) [3,4][3][4]. Congenital forms of hyperinsulinism (CHI) can be divided into the following two main groups: channelopathies defects in ion channels located on β-cells membrane and metabolopathies defects in the metabolic pathways leading to increased insulin release. The most common mutations are those affecting the KATP channel genes–ABCC8 (36.8%) and KCNJ11 (5.9%) (Figure 1) [5]. Some of the cases of HH are associated with genetic syndromes; Beckwith–Wiedemann syndrome is the most common [6]. The incidence of CHI is estimated at 1:50,000 live births, but in some populations, such as Finland or Saudi Arabia, the incidence reaches from 1:2500 to 1:3000 live births [7]. Transient forms of HH are secondary to conditions such as perinatal asphyxia, intra-uterine growth retardation, erythroblastosis fetalis, maternal diabetes mellitus or the administration of intravenous glucose during labor [6].
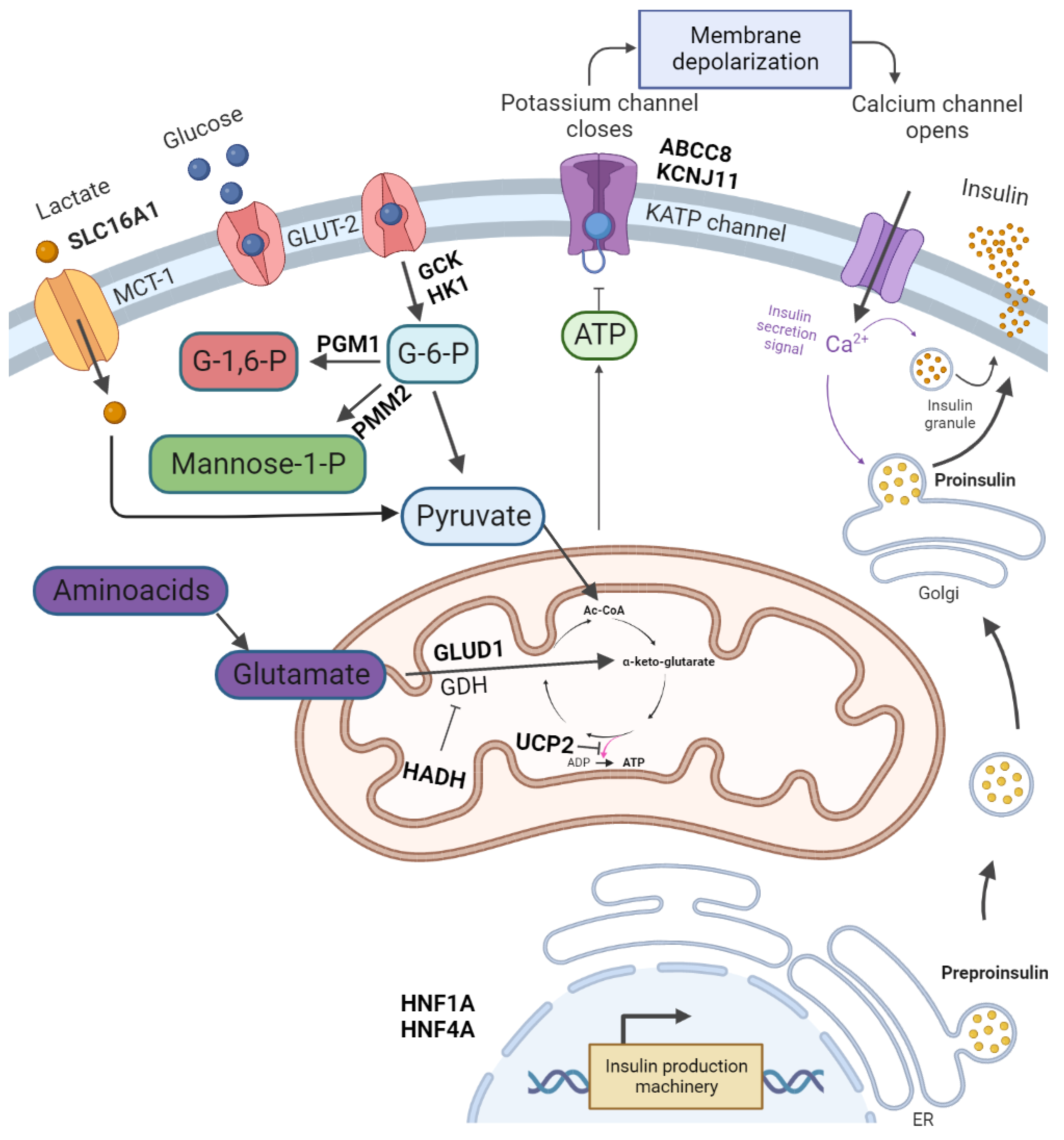
Figure 1. The regulation of insulin release and gene mutations involved in CHI pathogenesis. Glucose enters pancreatic β-cell via glucose transporter 2 (GLUT-2). It is then phosphorylated by glucokinase. The product of glycolysis–pyruvate is a substrate of the tricarboxylic acid cycle in mitochondria. The tricarboxylic acid cycle product is ATP. An increase in ATP levels leads to a sequence of KATP channel closure, membrane depolarisation, opening of voltage-dependent Ca2+ channels, Ca2+ influx and eventually insulin exocytosis. Glutamate dehydrogenase (GDH) catalyses the oxidative deamination of glutamate to alpha-ketoglutarate and ammonia. Further processes lead to amino acid-induced insulin secretion. PGM1 and PMM2 are responsible for glycosylation. Monocarboxylate transporter 1 (MCT1) catalyses the transport of monocarboxylates–pyruvate and lactate. Enhanced MCT1 expression increases pyruvate metabolism in the tricarboxylic acid cycle. Created with BioRender.com, accessed on 22 September 2022.
Table 1.
Genes in CHI pathogenesis.
| Gene | Full Gene Name | Diazoxide Responsiveness |
|---|---|---|
| ABCC8 | ATP-Binding Cassette Subfamily C Member 8 | Yes/No |
| KCNJ11 | Potassium Inwardly-Rectifying Channel Subfamily J Member 11 | No |
| GLUD1 | Glutamate Dehydrogenase 1 | Yes |
| GCK | Glucokinase | Yes/No |
| HADH | Hydroxyacyl-CoA Dehydrogenase | Yes |
| SLC16A1 (MCT-1) | Solute Carrier Family 16 Member 1 (Monocarboxylate Transporter Subtype 1) | Yes/No |
| UCP2 | Uncoupling Protein 2 | Yes |
| HNF4A | Hepatocyte Nuclear Factor 4A | Yes |
| HNF1A | Hepatocyte Nuclear Factor 1A | Yes |
| HK1 | Hexokinase 1 | Yes |
| PGM1 | Phosphoglucomutase 1 | No |
| PMM2 | Phosphomannomutase 2 | Yes |
2. Pathophysiology and Symptoms of CHI
The KATP channel reacts to an increased concentration of ATP (caused by glucose metabolism) by closure of the channel. This leads to β-cell membrane depolarisation and opening of the voltage-dependent Ca2+ channel. The increase in intracellular Ca2+ triggers insulin secretion [8]. The KATP channel consists of four sulphonylurea receptor 1 (SUR1) subunits, encoded by ABCC8, and four inward-rectifier potassium channel (Kir6.2) subunits, encoded by KCNJ11 [9]. The most severe forms of HH are caused by an inactivating mutation of the ABCC9 and KCNJ11 genes, which together are responsible for 36–70% of CHI cases [10,11][10][11].
Insulin decreases blood glucose concentrations by driving it into the insulin-sensitive tissues and inhibiting gluconeogenesis and glycolysis [2]. Insulin also inhibits lipolysis and ketone body synthesis. Therefore, in hyperinsulinaemic hypoglycaemia, the brain cannot be supplied with alternative energy sources. The combination of hypoketonaemic hypoglycaemia may lead to brain damage [5]. CHI is symptomatic mostly in early infancy and the neonatal period. Severe hypoglycaemia may even cause seizures or/and coma. The newborns are usually too big for their gestational age as a consequence of the high insulin level acting as a growth factor in utero [11]. Less specific symptoms of hypoglycaemia in the neonatal period include poor feeding, lethargy, irritability, apnoea and hypothermia [2]. In clinical examination, some newborns present cardiomyopathy and hepatomegaly [9].
3. Diagnosis of HH
An infant’s glucose blood concentration at birth amounts to 70% of the maternal levels. Within 1 h, it may decrease to 20–25 mg/dL. Then, over the next hours and days, it begins to rise [12]. The diagnostic criteria for patients with HH are based on the clinical presentation and biochemical profile. Plasma glucose levels of less than 54 mg/dL are accompanied by detectable serum insulin, detectable C-peptide, suppressed or low serum ketone bodies and suppressed or low serum concentrations of free fatty acids. The supportive evidence is a glucose infusion rate of >8 mg/kg per minute which is required to maintain normoglycaemia [9]. In the case of an unclear cause of hypoglycaemia, a glucagon test is performed. In this test, glucagon is administered intravenously at a dose of 0.03 mg/kg, and then blood glucose is measured at 0, 10, 20 and 30 min. CHI is diagnosed when the blood glucose level is higher than 30 mg/dL [9].
Histologically, CHI is divided into three types–diffuse, focal and atypical [13]. Focal lesions are responsible for 30–40% of congenital hyperinsulinaemic hypoglycaemia (CHH). Abnormal pancreatic cells are limited to a single location. The diffuse form of CHH accounts for 60–70% cases. The pathological β-cells are disseminated throughout the pancreas. The atypical form is described as a mosaic pattern of the two previous forms [6]. The gold standard in determining the form of HH is fluorine-18-dihydroxyphenyloalanine positron emission tomography ((18)F-DOPA PET). The sensitivity and specificity of (18)F-DOPA PET or PET/CT in differentiating between focal and diffuse CHI is 89% and 98%, respectively [14]. Rapid genetic testing of KATP channel genes allows for determining the histological subtype of the disease. It should be considered in diazoxide-unresponsive cases before performing (18)F-DOPA PET. The biallelic or single dominant KATP channel disease-causing variant confirms the diffuse form of HH, while a paternally inherited recessive KATP channel variant is linked to the focal form with a sensitivity of 97% [15,16,17][15][16][17].
Differential Diagnosis
The causes of neonatal hypoglycemia can be divided into four main groups: hyperinsulinism, inborn metabolic errors, counter-regulatory hormone deficiency and increased glucose requirement (Table 2) [18].
Table 2.
Main causes of neonatal hypoglycaemia.
| Transient neonatal hypoglycemia caused by hyperinsulinism |
|
| Patent neonatal hypoglycemia | Inborn metabolic errors (impaired gluconeogenesis):
|
Congenital hyperinsulinism:
|
|
Counter-regulatory hormone deficiency:
|
|
Increased glucose requirement:
|
The majority of neonatal hypoglycemia cases are caused by delayed metabolic adaptation after birth. Some patients require high glucose infusions for a week or more until they develop physiological metabolic processes [19].
Metabolic or hormonal etiology should be suspected in low-risk cases or unusual severity of hypoglycemia [19]. Most of these causes can be excluded or confirmed with laboratory testing [19,20,21,22,23][19][20][21][22][23] (Table 3). The critical samples for laboratory investigations should be obtained at the time of a hypoglycaemic episode [19].
Table 3. Suggested laboratory investigations of the critical samples (taken during a hypoglycaemic episode) for differential diagnosis of neonatal hypoglycaemia.
| Suggested Investigations in Differential Diagnosis |
|---|
| Complete blood count |
| Arterial blood gas |
| Blood glucose |
| Lactate |
| Pyruvate |
| Alanine |
| Glycerol |
| Ketone bodies |
| Plasma insulin |
| Free fatty acids |
| C-peptide |
| Plasma total and free carnitine, acylcarnitine profile |
| Ammonia |
| Electrolytes, blood urea nitrogen, creatinine |
| Uric acid |
| Growth hormone |
| Cortisol |
| Thyroid hormones |
| IGF-1 |
| Galactosaemia screen |
| Ketones and organic acids in urine |
References
- Senniappan, S.; Shanti, B.; James, C.; Hussain, K. Hyperinsulinaemic Hypoglycaemia: Genetic Mechanisms, Diagnosis and Management. J. Inherit. Metab. Dis. 2012, 35, 589–601.
- Roženková, K.; Güemes, M.; Shah, P.; Hussain, K. The Diagnosis and Management of Hyperinsulinaemic Hypoglycaemia. J. Clin. Res. Pediatr. Endocrinol. 2015, 7, 86.
- Galcheva, S.; Demirbilek, H.; Al-Khawaga, S.; Hussain, K. The Genetic and Molecular Mechanisms of Congenital Hyperinsulinism. Front. Endocrinol. 2019, 10, 111.
- Kostopoulou, E.; Shah, P. Hyperinsulinaemic Hypoglycaemia—An Overview of a Complex Clinical Condition. Eur. J. Pediatr. 2019, 178, 1151–1160.
- Rahman, S.A.; Nessa, A.; Hussain, K. Molecular Mechanisms of Congenital Hyperinsulinism. J. Mol. Endocrinol. 2015, 54, R119–R129.
- Gϋemes, M.; Rahman, S.A.; Kapoor, R.R.; Flanagan, S.; Houghton, J.A.L.; Misra, S.; Oliver, N.; Dattani, M.T.; Shah, P. Hyperinsulinemic Hypoglycemia in Children and Adolescents: Recent Advances in Understanding of Pathophysiology and Management. Rev. Endocr. Metab. Disord. 2020, 21, 577.
- Lord, K.; De León, D.D. Hyperinsulinism in the Neonate. Clin. Perinatol. 2018, 45, 61–74.
- Shimomura, K.; Maejima, Y. KATP Channel Mutations and Neonatal Diabetes. Intern. Med. 2017, 56, 2387.
- Shah, P.; Rahman, S.A.; Demirbilek, H.; Güemes, M.; Hussain, K. Hyperinsulinaemic Hypoglycaemia in Children and Adults. Lancet Diabetes Endocrinol. 2017, 5, 729–742.
- Takasawa, K.; Miyakawa, Y.; Saito, Y.; Adachi, E.; Shidei, T.; Sutani, A.; Gau, M.; Nakagawa, R.; Taki, A.; Kashimada, K.; et al. Marked Clinical Heterogeneity in Congenital Hyperinsulinism Due to a Novel Homozygous ABCC8 Mutation. Clin. Endocrinol. 2021, 94, 940–948.
- De Franco, E.; Saint-Martin, C.; Brusgaard, K.; Knight Johnson, A.E.; Aguilar-Bryan, L.; Bowman, P.; Arnoux, J.B.; Larsen, A.R.; May, S.; Greeley, S.A.W.; et al. Update of Variants Identified in the Pancreatic β-Cell KATP Channel Genes KCNJ11 and ABCC8 in Individuals with Congenital Hyperinsulinism and Diabetes. Hum. Mutat. 2020, 41, 884–905.
- Adamkin, D.H. Neonatal Hypoglycemia. Semin. Fetal Neonatal Med. 2017, 22, 36–41.
- Galcheva, S.; Al-Khawaga, S.; Hussain, K. Diagnosis and Management of Hyperinsulinaemic Hypoglycaemia. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 551–573.
- Treglia, G.; Mirk, P.; Giordano, A.; Rufini, V. Diagnostic Performance of Fluorine-18-Dihydroxyphenylalanine Positron Emission Tomography in Diagnosing and Localizing the Focal Form of Congenital Hyperinsulinism: A Meta-Analysis. Pediatr. Radiol. 2012, 42, 1372–1379.
- Hewat, T.I.; Johnson, M.B.; Flanagan, S.E. Congenital Hyperinsulinism: Current Laboratory-Based Approaches to the Genetic Diagnosis of a Heterogeneous Disease. Front. Endocrinol. 2022, 13, 1484.
- Snider, K.E.; Becker, S.; Boyajian, L.; Shyng, S.L.; MacMullen, C.; Hughes, N.; Ganapathy, K.; Bhatti, T.; Stanley, C.A.; Ganguly, A. Genotype and Phenotype Correlations in 417 Children with Congenital Hyperinsulinism. J. Clin. Endocrinol. Metab. 2013, 98, E355–E363.
- Mohnike, K.; Wieland, I.; Barthlen, W.; Vogelgesang, S.; Empting, S.; Mohnike, W.; Meissner, T.; Zenker, M. Clinical and Genetic Evaluation of Patients with KATP Channel Mutations from the German Registry for Congenital Hyperinsulinism. Horm. Res. Paediatr. 2014, 81, 156–168.
- Vain, N.E.; Chiarelli, F.; Palermo, T.; Isidro, S.; Mejía, R.; Aires, B.; Sanatorio Trinidad Palermo, H. Neonatal Hypoglycaemia: A Never-Ending Story? Neonatology 2021, 118, 522–529.
- Deshpande, S.; Platt, M.W. The Investigation and Management of Neonatal Hypoglycaemia. Semin. Fetal Neonatal Med. 2005, 10, 351–361.
- Thornton, P.S.; Stanley, C.A.; De Leon, D.D.; Harris, D.; Haymond, M.W.; Hussain, K.; Levitsky, L.L.; Murad, M.H.; Rozance, P.J.; Simmons, R.A.; et al. Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J. Pediatr. 2015, 167, 238–245.
- Adamkin, D.H. Metabolic Screening and Postnatal Glucose Homeostasis in the Newborn. Pediatr. Clin. 2015, 62, 385–409.
- Ghosh, A.; Banerjee, I.; Morris, A.A.M. Recognition, Assessment and Management of Hypoglycaemia in Childhood. Arch. Dis. Child. 2016, 101, 575–580.
- Douillard, C.; Mention, K.; Dobbelaere, D.; Wemeau, J.L.; Saudubray, J.M.; Vantyghem, M.C. Hypoglycaemia Related to Inherited Metabolic Diseases in Adults. Orphanet J. Rare Dis. 2012, 7, 26.
More