Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Benjamin Ory.
Head and neck squamous cell carcinoma (HNSCC) is the most common type (90%) of head and neck cancers, a group of biologically similar malignancies that affects the oral cavity (mouth), nasal cavity, pharynx, larynx, and paranasal sinuses. The p53 transcription factor family, which includes TP53 (TP53), p73 (TP73), and p63 (TP63), is a protein family that has a wide range of functions, ranging from embryonic development through to tumor suppression [1]. Unlike p53, p63 and p73 knockout (KO) mice showed abnormal epithelial development, with truncated limbs, missing lachrymal or salivary glands, and missing teeth and hair follicles.
- p53
- TGF
- metastasis
1. p53 Protein Family
The p53 transcription factor family, which includes TP53 (TP53), p73 (TP73), and p63 (TP63), is a protein family that has a wide range of functions, ranging from embryonic development through to tumor suppression [1]. Unlike p53, p63 and p73 knockout (KO) mice showed abnormal epithelial development, with truncated limbs, missing lachrymal or salivary glands, and missing teeth and hair follicles [2,3][2][3]. p63 and p73 knockout (KO) mice also showed defective neurological development, with congenital hydrocephalus, hippocampal dysgenesis, and chronic inflammation, in the case of p73 KO [4,5][4][5].
In response to various cellular stress conditions such as DNA damage, hypoxia, nucleotide imbalance, and others, the tumor suppressor p53 induces target genes that are involved in cell cycle arrest, apoptosis, and DNA repair [6]. Most human cancers have p53 inactivation due to direct mutation, deletion, or disruption of critical regulatory mechanisms that are required for proper p53 function. Mutations or deletion of the p63 and p73 transcription factors, on the other hand, are uncommon in cancer [7,8][7][8].
p53 Protein Family: Structure and Functions
All three p53 family members are very similar and present a high homology both at the genomic and protein levels. Each contains an N-terminal transactivation domain (TAD), a central DNA binding domain (DBD) through which they regulate both shared and distinct transcriptional targets [9], and an oligomerization domain (OD) [1]. In addition, p63 and p73, but not p53, can contain a long C-terminal that is mainly composed of a sterile alpha motif (SAM) domain and a transactivation inhibitory domain (TID). The SAM consists of four α-helices and a small helix which enables protein–protein interactions whereas TID is a region that inhibits the transcriptional activity of TA isoforms through inter- or intra-molecular association with their TAD [10,11][10][11] (Figure 1). The DBD exhibits more than 60% homology between the three proteins, suggesting that they can bind to similar sequences and transactivate the same promoters. Furthermore, the high conservation of OD suggests the possibility of homo- and hetero-oligomer formation between the p53 family protein partners.
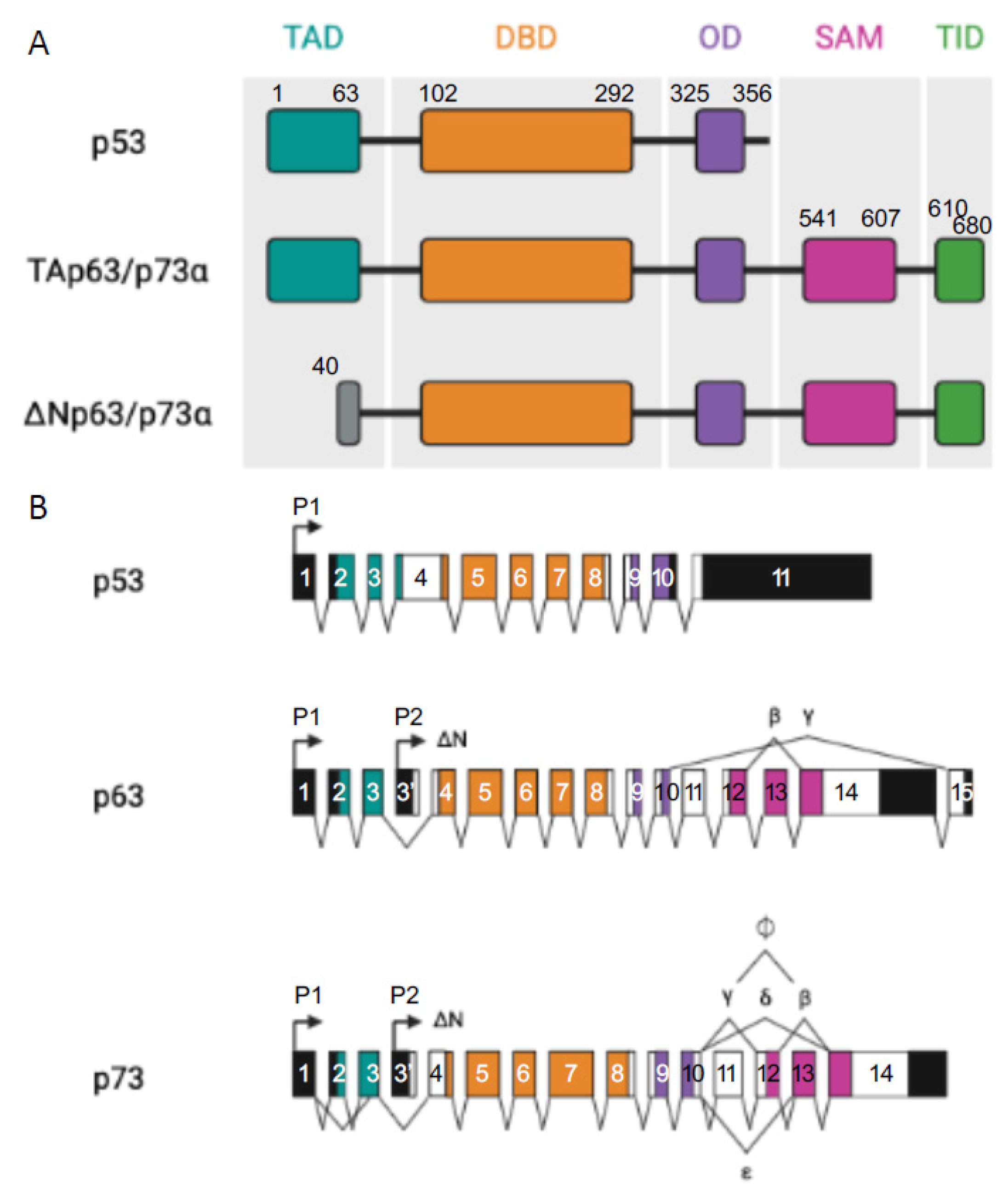
Figure 1. p53 family members. (A) Representation of the different domains in p53 and TA and ΔN p63α and p73α isoforms: transactivation domain (TA, in light grey), DNA binding domain (DBD, in red), oligomerization domain (OD, in yellow), sterile alpha domain (SAM, in dark grey), and the transactivation inhibitory domain (TID, in orange), introns (black lines). (B) Represents p53, p63, and p73 multiple spliced variants. P stands for different promoters, numbered boxes indicate exons, black boxes are untranslated sequences, and black lines are introns.
Moreover, p63 and p73 can be expressed from two distinct promoters (one upstream of exon 1 (P1) and another that is located within intron 3 (P2)) and can also be differentially spliced, thereby producing different isoforms. Transcription from the P1 promoter gives an N-terminal acidic TA domain (TAp63 and TAp73) whereas in the products that are transcribed from P2, this TA domain is absent (ΔNp63 and ΔNp73) [12].
From the C-terminal splicing of p63 and p73, a large variety of proteins can be generated. At least seven C-terminal isotypes have been identified for p73 (α, β, γ, δ, ε, ζ, and η) and three for p63 (α, β, and γ). In total, the p63 gene encodes for six different protein isoforms and the p73 gene expresses 35 mRNA variants that can theoretically encode for 28 different protein isoforms. Only 14 isoforms have been described so far [13]. Their contributions are not yet fully understood but some evidence points to the fact that these different C-termini could be involved in the capacity of TA isoforms to transactivate gene expression [7,8][7][8]. In both cases, p63α and p73α are the full-length isoforms (with SAM and TID present at their C-termini) and the others are the result of different truncations of those ones (Figure 1).
It has been shown that p53 also produces multiple isoforms through the use of two promoters and alternative mRNA splicing. Those isoforms are expressed in normal human tissues in a tissue-dependent manner [13].
Due to the high homology that is observed within the three members of the p53 protein, and the fact that, unlike the not functional p53, p63 and p73 are found overexpressed in the vast majority of cancers and their role in the malignant context has been questioned [14]. Whether p63 and p73 promote or not, human tumorigenesis and metastatic dissemination may depend on the predominant isoforms that are expressed in a specific tissue. TA isoforms have been demonstrated to transactivate distinct but overlapping subsets of known p53-regulated genes that are involved in cell-cycle arrest and apoptosis, as well as some other gene sets not regulated by p53. In contrast, the TAD truncated isoforms, ΔNp63 and ΔNp73 proteins, function as dominant-negative inhibitors of the p53 family, so that, these isoforms may be most likely related with protooncogenic functions [15,16][15][16]. Those conclusions come from studies indicating that TAp73 mediates apoptosis as a result of its nuclear accumulation following chemotherapy-induced DNA damage [17]. The overexpression of ΔNp63 and ΔNp73, in contrast, inhibits the pro-apoptotic effect of Tap73 in human tumors.
2. p63 and p73 Interactions in the Head and Neck Squamous Cell Carcinoma Model (HNSCC)
Head and neck squamous cell carcinoma (HNSCC) is the most common type (90%) of head and neck cancers, a group of biologically similar malignancies that affects the oral cavity (mouth), nasal cavity, pharynx, larynx, and paranasal sinuses. It arises from epithelial cells that line the mucosal surfaces of the head and the neck [18]. HNSCC is the sixth most common diagnosed cancer worldwide with 560,000 new cases and 300,000 deaths annually reported [19,20][19][20]. Although cervical lymph nodes are the main metastatic sites, the risk of dissemination is not so high and it depends on both the stage and location of the primary tumor [21]. Nevertheless, head and neck cancers are aggressive in nature. Tobacco and alcohol consumption are the main factors that are responsible for HNSCC apparition but some studies also point their initiation from the human papillomavirus (HPV) infection. Therapy is mainly based on surgery or radiotherapy at early stages whereas a combination of surgery, radiotherapy, and chemotherapy is applied in advanced stages, resulting in multiple toxic side effects [18].
HNSCC presents mutated p53, as it is in the majority of cancers, with mutations that are found in more than half of HNSCC malignancies. Rather than being mutated, p73 is overexpressed in a wide range of tumor types, including breast, lung, colon, and stomach cancers, as well as epithelial cancers such as HNSCC, with TAp73 being the most common isoform [22]. The P63 gene region (chromosome 3q27-28) is frequently amplified in squamous cell carcinomas [23,24][23][24] and its consequent protein overexpression is seen in up to 80% of HNSCCs. In these types of cancers, Np63 is the most prevalent isoform. ΔNp63α is the most predominant isoform in these kinds of malignancies [25,26][25][26]. As both partners possess homologous ODs, they are susceptible to interact and form homo- and hetero-oligomers (Figure 2). When p63 is expressed in cancer cells, homodimers between two ΔNp63α molecules (the most common expressed isoform) will be formed and will repress some apoptotic promoters as Puma and Noxa, promoting survival. In contrast, when p63 is absent, TAp73 is the most usual isoform which will form dimers as well, but this time promoting the expression of apoptotic genes.
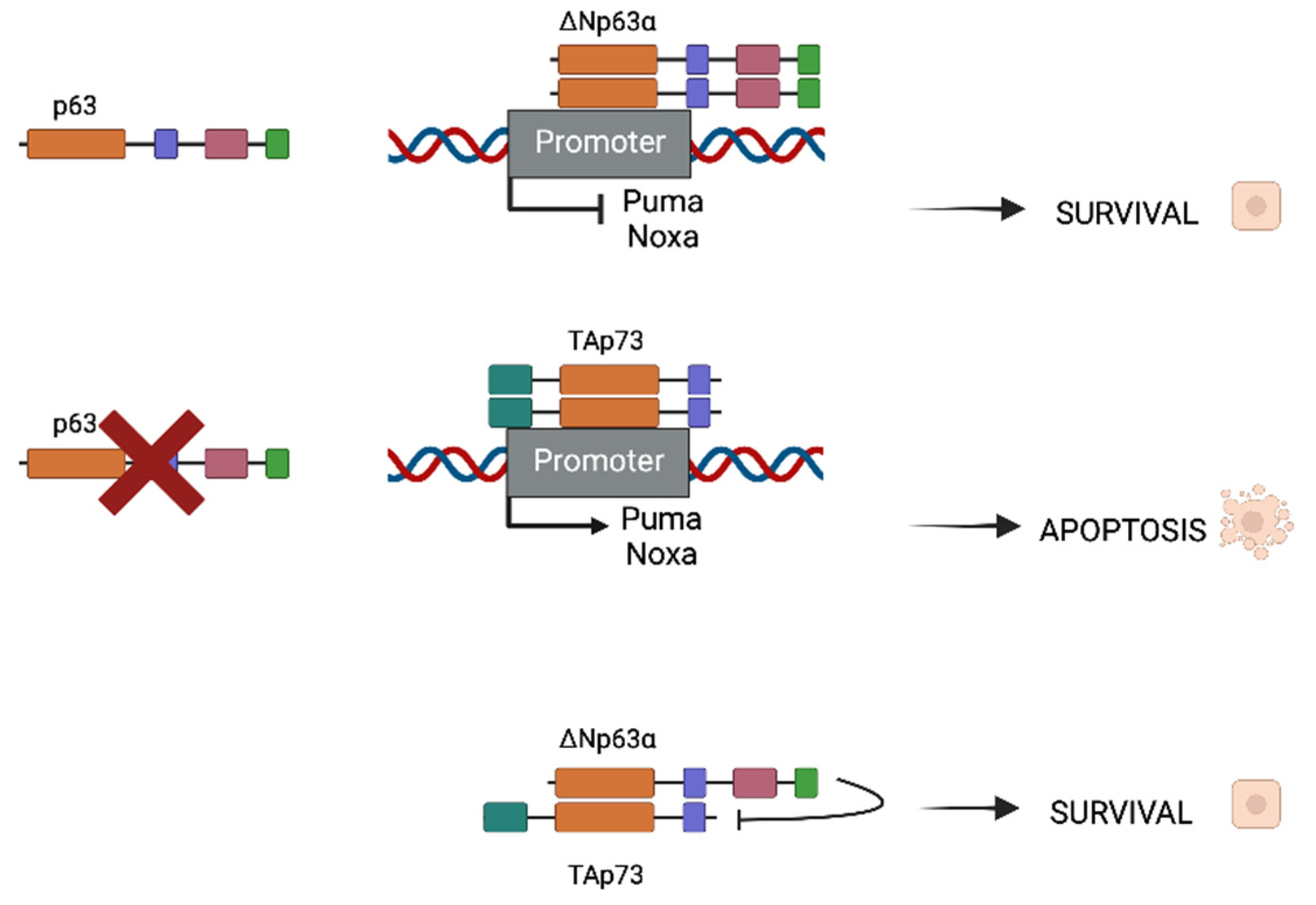
Figure 2. Different oligomerization of ΔNp63α and TAp73 depending on p63 presence or absence. When ΔNp63α is present in cancer cells, survival will be induced. In contrast, when p63 is not present, homodimers of TAp73 will be formed and will promote pro-apoptotic gene expression (Puma and Noxa for instance). In HNSCC, both ΔNp63α and TAp73 are expressed but TID of ΔNp63α is capable of inhibiting pro-apoptotic function of the TA isoform, thus promoting survival and cancer progression.
In the case where both populations coexist, heterodimers between ΔNp63α and TAp73 can be formed and are crucial for tumor maintenance. There is a balance that is established between both isoforms that can be destabilized if one of the isoforms surpasses the other. Depending on whether the isoform is more frequent, this disturbance of balance might result in improved survival or apoptotic characteristics. Physical interactions between p63 and p73 have been shown to be significantly stronger than homodimers, which is interesting. The main isoform in the HNSCC environment is ΔNp63, which results in Tap73-Np63 heterodimers. The TID of the dominant protein inhibits the TA isoform’s transcriptional activity, resulting in a survival phenotype.
The above-described studies emphasize the fact that HNSCC is a good model to better understand the involvement of p63 and p73 in cancer, thanks to its inactive p53. Several teams have used it to assess the implications of p63 (ΔNp63α) and p73 (TAp73β) in the cancer metastatic dissemination, in particular through the regulation of miRNA networks [27,28,29][27][28][29].
References
- Levrero, M.; De Laurenzi, V.; Costanzo, A.; Gong, J.; Wang, J.Y.; Melino, G. The p53/p63/p73 family of transcription factors: Overlapping and distinct functions. J. Cell Sci. 2000, 113 Pt 10, 1661–1670.
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713.
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718.
- Pozniak, C.D.; Radinovic, S.; Yang, A.; McKeon, F.; Kaplan, D.R.; Miller, F.D. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 2000, 289, 304–306.
- Yang, A.; McKeon, F. P63 and P73: P53 mimics, menaces and more. Nat. Rev. Mol. Cell Biol. 2000, 1, 199–207.
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310.
- Moll, U.M.; Slade, N. p63 and p73: Roles in development and tumor formation. Mol. Cancer Res. MCR 2004, 2, 371–386.
- Deyoung, M.P.; Ellisen, L.W. p63 and p73 in human cancer: Defining the network. Oncogene 2007, 26, 5169–5183.
- Leong, C.O.; Vidnovic, N.; DeYoung, M.P.; Sgroi, D.; Ellisen, L.W. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J. Clin. Investig. 2007, 117, 1370–1380.
- Chi, S.W.; Ayed, A.; Arrowsmith, C.H. Solution structure of a conserved C-terminal domain of p73 with structural homology to the SAM domain. EMBO J. 1999, 18, 4438–4445.
- Serber, Z.; Lai, H.C.; Yang, A.; Ou, H.D.; Sigal, M.S.; Kelly, A.E.; Darimont, B.D.; Duijf, P.H.; Van Bokhoven, H.; McKeon, F.; et al. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol. Cell. Biol. 2002, 22, 8601–8611.
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dotsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316.
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137.
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887.
- Melino, G.; De Laurenzi, V.; Vousden, K.H. p73: Friend or foe in tumorigenesis. Nat. Rev. Cancer 2002, 2, 605–615.
- Dohn, M.; Zhang, S.; Chen, X. p63alpha and DeltaNp63alpha can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene 2001, 20, 3193–3205.
- Gong, J.G.; Costanzo, A.; Yang, H.Q.; Melino, G.; Kaelin, W.G., Jr.; Levrero, M.; Wang, J.Y. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 1999, 399, 806–809.
- Suh, Y.; Amelio, I.; Guerrero Urbano, T.; Tavassoli, M. Clinical update on cancer: Molecular oncology of head and neck cancer. Cell Death Dis. 2014, 5, e1018.
- Safdari, Y.; Khalili, M.; Farajnia, S.; Asgharzadeh, M.; Yazdani, Y.; Sadeghi, M. Recent advances in head and neck squamous cell carcinoma—A review. Clin. Biochem. 2014, 47, 1195–1202.
- Parkin, D.M.; Pisani, P.; Ferlay, J. Estimates of the worldwide incidence of 25 major cancers in 1990. Int. J. Cancer 1999, 80, 827–841.
- Vokes, E.E.; Weichselbaum, R.R.; Lippman, S.M.; Hong, W.K. Head and neck cancer. N. Engl. J. Med. 1993, 328, 184–194.
- DeYoung, M.P.; Johannessen, C.M.; Leong, C.O.; Faquin, W.; Rocco, J.W.; Ellisen, L.W. Tumor-specific p73 up-regulation mediates p63 dependence in squamous cell carcinoma. Cancer Res. 2006, 66, 9362–9368.
- Hibi, K.; Trink, B.; Patturajan, M.; Westra, W.H.; Caballero, O.L.; Hill, D.E.; Ratovitski, E.A.; Jen, J.; Sidransky, D. AIS is an oncogene amplified in squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2000, 97, 5462–5467.
- Bjorkqvist, A.M.; Husgafvel-Pursiainen, K.; Anttila, S.; Karjalainen, A.; Tammilehto, L.; Mattson, K.; Vainio, H.; Knuutila, S. DNA gains in 3q occur frequently in squamous cell carcinoma of the lung, but not in adenocarcinoma. Genes Chromosom. Cancer 1998, 22, 79–82.
- Rocco, J.W.; Leong, C.O.; Kuperwasser, N.; DeYoung, M.P.; Ellisen, L.W. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 2006, 9, 45–56.
- Sniezek, J.C.; Matheny, K.E.; Westfall, M.D.; Pietenpol, J.A. Dominant negative p63 isoform expression in head and neck squamous cell carcinoma. Laryngoscope 2004, 114, 2063–2072.
- Rodriguez Calleja, L.; Jacques, C.; Lamoureux, F.; Baud’huin, M.; Tellez Gabriel, M.; Quillard, T.; Sahay, D.; Perrot, P.; Amiaud, J.; Charrier, C.; et al. DeltaNp63alpha Silences a miRNA Program to Aberrantly Initiate a Wound-Healing Program That Promotes TGFbeta-Induced Metastasis. Cancer Res. 2016, 76, 3236–3251.
- Kakuki, T.; Kurose, M.; Takano, K.; Kondoh, A.; Obata, K.; Nomura, K.; Miyata, R.; Kaneko, Y.; Konno, T.; Takahashi, S.; et al. Dysregulation of junctional adhesion molecule-A via p63/GATA-3 in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 33887–33900.
- Dong, J.; Li, J.; Li, Y.; Ma, Z.; Yu, Y.; Wang, C.Y. Transcriptional super-enhancers control cancer stemness and metastasis genes in squamous cell carcinoma. Nat. Commun. 2021, 12, 3974.
More