Cancer癌症是全球第二大死因,并且是一种遗传性疾病,具有高度遗传性。全基因组关联研究 ranks as the second leading cause of death worldwide, and, being a genetic disease, it is highly heritable. Over the past few decades, genome-wide association studies (GWAS) have identified many risk-associated loci harboring hundreds of single nucleotide polymorphisms (SNPs). Some of these cancer-associated SNPs have been revealed as causal, and the functional characterization of the mechanisms underlying the cancer risk association has been illuminated in some instances.已经确定了许多风险相关位点,其中包含数百个单核苷酸多态性 (SNP)。其中一些与癌症相关的 SNP 已被揭示为因果关系,并且在某些情况下阐明了癌症风险关联机制的功能特征。
1. Functional Mechanisms of Coding Region SNPs编码区 SNP 的功能机制
位于编码区的S
ingle nucleotide polymorphisms (SNP
s) located in coding regions can be divided into two types: synonymous and non-synonymous mutations. Although synonymous mutations do not affect the amino acid sequence of the protein, they may change the expression of the protein by affecting post-transcriptional modifications, translation rates, and other processes. In contrast, non-synonymous SNPs (nsSNPs) cause the substitution of amino acids, thereby resulting in changes to the protein structure, its physical and chemical properties (stability, solubility, etc.), and its function. At present, there are many biological software packages (such as SIFT(可分为两类:同义突变和非同义突变。虽然同义突变不影响蛋白质的氨基酸序列,但它们可能通过影响转录后修饰、翻译率和其他过程来改变蛋白质的表达。相反,非同义单核苷酸多态性(nsSNPs)引起氨基酸的取代,从而导致蛋白质结构、物理和化学性质(稳定性、溶解性等)及其功能发生变化。目前,有许多生物学软件包(如 SIFT(Sorting Intolerant From Tolerant
), F-SNP(the Functional Single Nucleotide Polymorphism) and PolyPhen) that can be used to predict the effect of nsSNPs on protein)、F-SNP(功能单核苷酸多态性)和 PolyPhen)可用于预测 nsSNPs 对蛋白质结构和功能的影响 [ structure41 and, function42, [1][2][3].43 Compared]。与位于基因非编码区的 to SNPs located in gene non-coding regions, the functional mechanism underlying tumor-associated nsSNPs is relativelySNP 相比,肿瘤相关 nsSNP 的功能机制相对简单 [ simple44 [4]. Combined with whole exon]。结合全外显子分析,已确定多个编码区 analysis, several coding region SNP
s have been identified to be associated with colorectal cancer development. For example, the missense mutation rs 与结直肠癌的发展相关。例如,位于 SH2B3 结构域的错义突变 rs3184504 (p. trp263ARg)
located in a domain of SH2B3 may change the function of the protein in the context of regulating cell division. Other coding variants may also affect variable shear (可能会在调节细胞分裂的背景下改变蛋白质的功能。其他编码变体也可能影响可变剪切(RS16888728
, UTP23)、UTP23)[ [5].45 The]。位于基因编码区内的 mechanism by which SNPs locatedSNP 影响疾病风险的机制与所产生的编码蛋白的功能密不可分。
一些风险位点对产生的蛋白质的氨基酸序列产生影响。示例包括肺癌 [ 46 within] the coding和乳腺癌 [ regions47 of]中的 genes affect the risk of disease is inseparable from the function of the resulting coded proteins.
Some risk loci exert an effect on the amino acid sequence of the produced protein. Examples include BRCA2 p.Lys3326Ter (rs11571833)
, and和 CHEK2 p.Ile157Thr (rs17879961)
in lung [6] and。假定对此类变体的机械解释相对简单。除了上述之外,编码 breast [7] cancers. The mechanistic interpretation of such variants is presumed to be relatively simple. In addition to the aforesaid, coding SNP
s can affect RNA processing; an example is还会影响 RNA 加工;一个例子是 TP53 的 3' 非翻译区中的 rs78378222
in the 3′ untranslated region of TP53, whereby the risk-corrected variation alters the sequence ,风险校正变异将序列 AATAAA
to更改为 AATACA
, thereby changing the polyadenylation signal of TP53, and ultimately resulting in the impaired 3′-end processing of ,从而改变 TP53 的聚腺苷酸化信号,并最终导致 TP53 mRNA
[8][9].的 Some3'-末端加工受损 variants[ can48 also, affect splicing49].
Tian and his colleagues identified a single-nucleotide variation in the 一些变异也会影响剪接。Tian 和他的同事在ELP2基因中发现了一个单核苷酸变异,它通过剪接数量性状基因座 gene that affect ELP2 exon pre-mRNA splicing through splicing a quantitative trait locus (sQTL)
影响ELP2外显子前体 mRNA 剪接 [ 50 [10].
Researchers have]。
研究人员通常专注于感兴趣的特定信号通路、基因和基因修饰,同时还进行全外显子关联分析 often focused on specific signaling pathways, genes, and genetic modifications of interest, while also performing whole-exon association analysis (GWAS)
to find any relevant coding SNPs with large effects on these molecules and processes. For example, Li and colleagues used exon sequencing and conducted an association analysis of 12 important genes involved in 以发现对这些分子和过程有重大影响的任何相关编码 SNP。例如,Li 及其同事使用外显子测序并对参与 TGF-β
signaling to find that low-frequency causative variation in the 信号传导的 12 个重要基因进行了关联分析,发现 TGF-β
pathway contributes to colorectal carcinoma 通路中的低频致病变异有助于结直肠癌 (CRC)
susceptibility. They discovered that the missense variation r易感性。他们发现位于 SMAD7 基因中的错义变异 rs3764482 (c. 83C>T; p. S28F
) located in the gene SMAD7 was consistently and)始终与 strongly associated with CRC
risk. The 风险密切相关。与显性等位基因 C 相比,rs3764482
allele T was more effective compared to the dominant allele C in limiting 等位基因 T 在限制 TGF-β
signaling and reducing the phosphorylation of receptor-regulated 信号传导和通过阻碍下游基因的激活减少受体调节的 SMADs (R-SMADs)
via impeding the activation of downstream genes, thereby promoting cancer cell proliferation and contributing to CRC pathogenesis的磷酸化方面更有效,从而促进癌细胞增殖并促进 CRC发病机制 [ 51 [11].
Coding]。
编码 SNP
s may also affect gene and protein modifications. The N6-methyladenosine也可能影响基因和蛋白质修饰。N6-甲基腺苷 (m6A)
modification is critical for ensuring messenger RNA stability and is involved in many biological activities, including pre-mRNA splicing, 3’-end processing, nuclear export, translation regulation, mRNA degradation, and the DNA damage修饰对于确保信使 RNA 的稳定性至关重要,并参与许多生物活动,包括前 mRNA 剪接、3' 端加工、核输出、翻译调节、mRNA 降解和 DNA 损伤反应 [ response52 [12][13]., The53 ]。m6A
methylation modification occurs in the messenger RNA甲基化修饰发生在信使 RNA (mRNA)
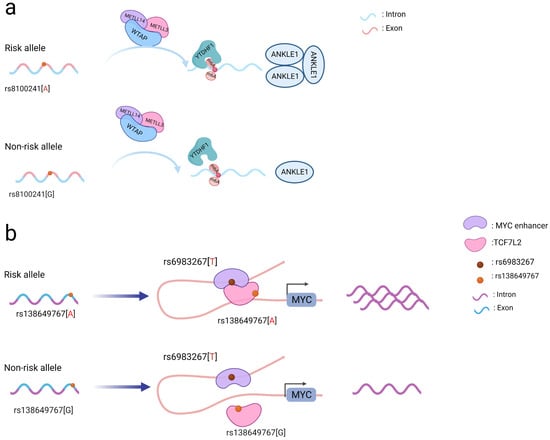
and can be formed by methylation “writers” and removed by demethylation “erasers”中,可由甲基化“写入器”形成,并由去甲基化“擦除器”去除 [14].[ Rs8100241,54 located]。位于 in the gene ANKLEANKLE1 基因中的Rs8100241,被鉴定为与 was identified to be associated with susceptibility to both CRC and breast cancer. The presence of the rsCRC 和乳腺癌的易感性相关。rs8100241
risk allele A (Figure风险等位基因 A 的存在(图 1a)
combined与 with the m6A “writer” complex (comprised of the proteins m6A“编写器”复合体(由蛋白质 METTL3
, 、METTL14
, and WTAP) and the m6A “reader” protein 和 WTAP 组成)和 m6A“读取器”蛋白 (YTHDF1)
was found to increase the levels of the m6A modification on the gene 结合,被发现可增加基因ANKLE1上的 and consequently increase its protein expression. Mechanistically, m6A 修饰水平,从而增加其蛋白表达。从机制上讲,ANKLE1
functions as a potential tumor suppressor by decreasing CRC cell proliferation通过减少 CRC 细胞增殖同时保持基因组完整性,作为潜在的肿瘤抑制因子发挥作用,从而有助于降低 CRC 的风险 [ while55 maintaining genomic integrity, thereby contributing to a lower risk of CRC [15].
]。
Figure图 1.
SNP 编码所采用的作用机制示意图。( a ) Schematic diagram of the action mechanism employed by coding SNPs. (a) The A allele of the rs8100241 variant, which is found in the ANKLE1 second exon region, has been linked to a lower risk of CRC by increasing ANKLE1 mRNA m6A levels and thus facilitating ANKLE1 protein expression, thereby potentially functioning as a negative regulator to hinder cell growth by maintaining genomic stability. (
在ANKLE1第二外显子区域发现的 rs8100241 变体的 A 等位基因通过增加ANKLE1 mRNA m6A 水平并因此促进 ANKLE1 蛋白表达而与较低的 CRC 风险相关联,从而可能起到负调节剂的作用通过维持基因组稳定性来阻碍细胞生长。( b) Interaction between the TCF7L2 missense variant rs138649767 and a regulatory variant rs6983267 in the MYC enhancer and promoter on the expression of MYC.
) MYC增强子和启动子中TCF7L2错义变体 rs138649767 和调节变体 rs6983267之间的相互作用对 MYC表达的影响。
Notably, coding SNPs may interact with other SNPs to produce a stronger functional role [5]. The rs138649767 A allele (Figure 1b) located in the exon region of
值得注意的是,编码 SNP 可能与其他 SNP 相互作用以产生更强的功能作用 [ 45 ]。位于TCF7L2 can activate the
外显子区的rs138649767 A等位基因(图1b)可激活含有rs6983267等位基因G的MYC enhancer containing rs6983267 allele G to promote the expression of MYC [16]. SNPs occurring in the exons and introns of
增强子,促进MYC的表达[ 56 ]。SMAD7 may affect its regulation and jointly affect downstream signaling pathways involving SMAD7 and TGFβ [11]. As a result, while examining SNPs in coding regions, the interactions between them should be taken into account to better understand their functional processes.
外显子和内含子中出现的SNP可能会影响其调控,并共同影响涉及 SMAD7 和 TGFβ 的下游信号通路 [ 51 ]。因此,在检查编码区中的 SNP 时,应考虑它们之间的相互作用,以更好地了解它们的功能过程。
2. Functional Mechanisms of Non-Coding Region SNPs 非编码区 SNP 的功能机制
Accumulating evidence shows that a SNP in non-coding regions is the most common type of genetic variation in the human genome, accounting for 90% of inter-individual variation [17][18]. Depending on the location, the region can harbor a response element that is either proximal (promoter, enhancer, or super-enhancer) or distal (intergenic or intra-genic). The risk loci identified by GWAS were located in the genomic regions of cell type-specific active chromatin, and most of them were quantitative trait loci, methylation quantitative trait loci and transcription factor (TF) binding related loci. Chromatin conformational studies have helped to link regulatory regions localized by SNPs to their respective target genes [17][19][20]. These loci may be involved in gene transcription, post-transcriptional processing, translation, post-translational modifications, and other processes to regulate gene expression. Many target genes have been identified using expression quantitative trait loci (eQTL) to detect the relationship between SNPs and gene expression. Non-coding SNPs can regulate the transcription of target genes by sequence-proximal (cis)- or distal (trans)-interactions. Studies have found that histone modifications in the regions of such risk SNPs are particularly abundant, especially those related to promoter and enhancer activities (H3K4me3, H3K4me1, H3K27ac). Most SNPs are predicted to destroy the binding motifs of specific transcription factors. For example, rs6983267 may change the binding of transcription factors such as MYC, CTCF, and TCF7L2 [21]. In addition to affecting gene transcription levels by altering transcription factor-binding sites (TFBS), non-coding SNPs also change epigenetic modifications and/or the chromatin structure to influence target gene expression. Through the above method, non-coding SNP participates in cell proliferation, apoptosis, migration, and invasion.
2.1. Genetic Variants That Alter Promoters
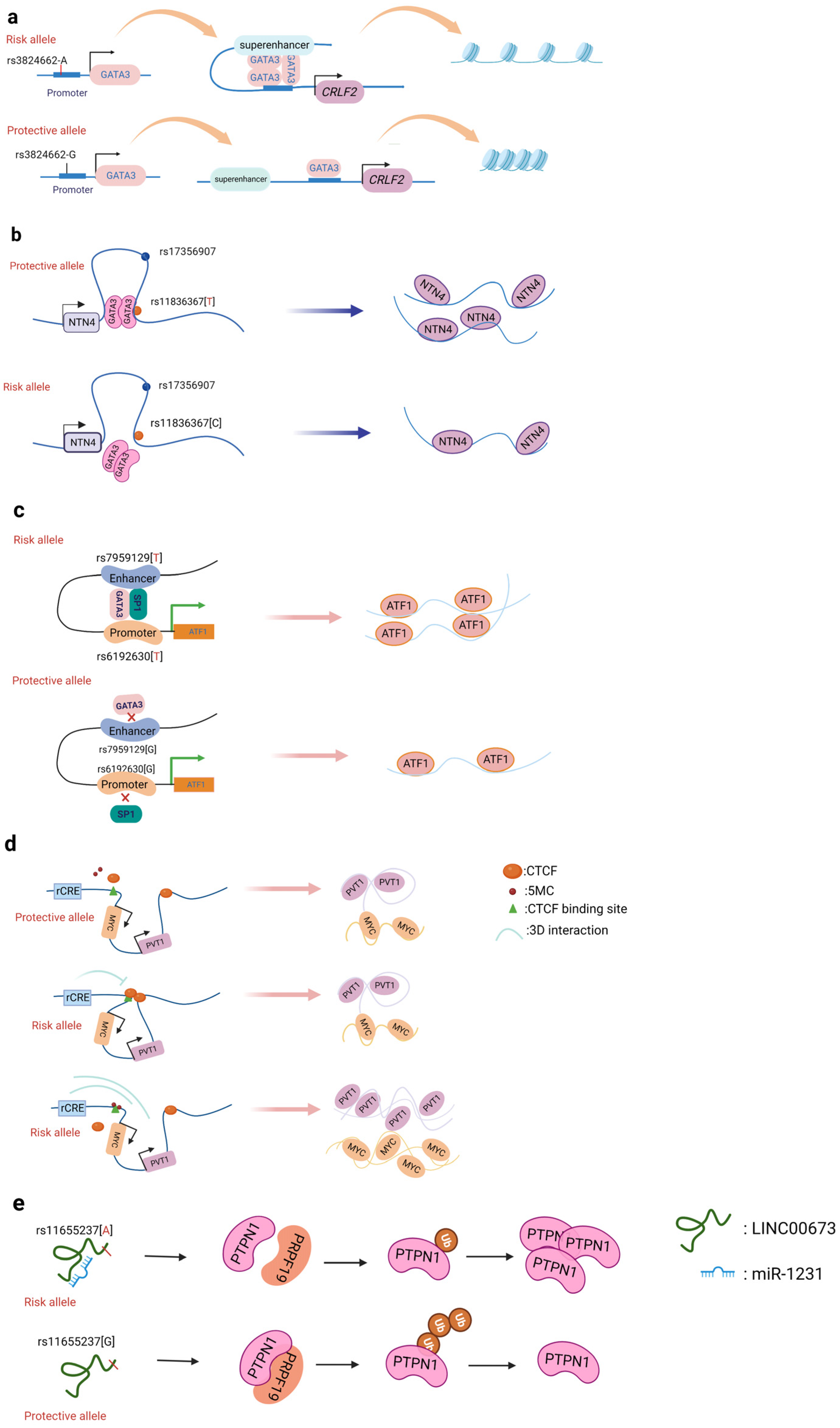
A promoter is a sequence of DNA that is recognized, bound, and serves to initiate transcription by RNA polymerase. Promoters contain variations of a conserved sequence required for the specific binding of RNA polymerase and transcription initiation. Most promoters are located upstream of the transcription initiation point of structural genes, and the promoter itself is not transcribed [22]. Promoters are located upstream of the 5’ end of a given structural gene, and they activate RNA polymerase to bind accurately to the template DNA with specificity for inducing the initiation of transcription [22]. Promoters do not control gene activity themselves; rather, gene activity is regulated by binding to proteins called transcription factors (TF). SNPs within promoter regions generally play a regulatory role by influencing the binding of such transcription factors. A recently reported example is that of the SNP rs13278062 located in the promoter of death receptor 4 (DR4) which confers an altered risk of colorectal cancer. The study revealed that the rs13278062 G>T variant changed the binding affinity of the transcription factor Sp1/NF1, increased the expression of DR4, and thus suppressed carcinogenesis and metastasis of colorectal cancer [23]. The MPO promoter SNP rs2333227 increases the malignant characteristics of colorectal cancer by changing the promoter’s affinity to AP-2α [24]. The variant SNP rs10993994 located in the upstream promoter of the gene MSMB is also found to be overrepresented in individuals with prostate cancer; this is attributed to stronger CREB binding and thus increased promoter activity [25]. Furthermore, the SNP rs11672691 is a risk locus associated with prostate cancer that is related to the lncRNA PCAT19. The non-risk variant rs11672691 and its linkage disequilibrium (LD) SNP rs887391 are more likely to bind the TFs NKX3.1 and YY1 to the PCAT19-short promoter, thereby leading to increased promoter but lower enhancer activity, which then activates PCAT19-short, and ultimately results in lower prostate cancer susceptibility [26]. SNPs in promoter regions of multiple genes, including TERT, KLHDC7A, PIDD1, and ESR1, have been discovered in breast cancer by GWAS, with reporter studies revealing that independent risk alleles change target promoter activity [27][28]. Most of the reported promoter changes exert their regulatory effects by altering TF binding. The SNP rs3824662 allele A (Figure 2a) increases chromatin accessibility by changing the TF GATA3 expression, promoting the binding of GATA3 with the CRLF promoter, and ultimately forming a chromatin loop [29].
Figure 2. Schematic diagram of the action mechanism employed by non-coding SNPs. (a) The SNP rs3824662 allele A increases chromatin accessibility by inducing GATA3 expression, promoting the binding of GATA3 with the CRLF promoter, and ultimately forming a chromatin loop. (b) The NTN4 enhancer risk variant rs11836367 binds to the TF GATA3 to regulate NTN4 expression, ultimately promoting breast carcinoma initiation and progression. (c) Enhancer SNP rs7959129 risk allele G interacts with promoter SNP rs6192603 risk allele G contributing to ATF1 expression by binding TFs GATA3 and SP1. (d) The risk allele rs11986220 and higher methylation at –10 Kb synergistically function to confer a greater risk of tumor; however, when -20 Kb is hypomethylated, the function of the risk SNP is inhibited by the enhancer-blocking insulator loop mediated by CTCF. (e) The risk variant rs11655237 in LINC00673 creates a miR-1231–binding site that interferes with the expression of LINC00673 and contributes to pancreatic cancer susceptibility.
2.2. Genetic Variants That Alter Enhancers
Enhancers are regions of DNA sequence that can increase the cis-acting transcription of their target gene sequences. Enhancers each differ in their distance from their target promoter(s); in mammalian species, an enhancer can be 100 bp to Mb away from their target gene [30]. Enhancers, unlike promoters, can be found anywhere in a gene; they can be positioned either upstream or downstream of their target genes, or even within another gene’s gene body, and enhancer regulation can circumvent other genes irrespective of their orientation. Enhancers must bind to specific protein factors to enhance the transcription of their target. Enhancers generally have tissue or cell specificity, whereby they only show activity in certain cells or tissues, which is determined by the specific protein factors present in these cells or tissues [31]. Enhancers are typically recognized by the epigenetic marks H3K4me1 and H3K27ac, which are present in active enhancer elements. Conversely, H3K27me3 is regarded as a silent epigenetic mark associated with lower enhancer activity [32][33]. GWAS-identified risk loci for common illnesses are often found in non-coding areas, and many of these are thought to function as enhancers [34]. According to emerging data, these SNPs may influence gene regulation by changing the binding of important TFs to critical transcriptional enhancers [35].
2.2.1. Breast Cancer
Of all cancers, breast cancer has so far yielded the greatest number of discovered risk loci [36]. Understanding the driving mechanism(s) underlying malignant transformation provides the prospect of combating cancer recurrence and treatment resistance. Zhang et al. identified that the SNP rs4971059 resides in the sixth intron and within an active enhancer element of the TRIM46 gene. By using CRISPR/Cas9-mediated homologous recombination, they constructed the SNP rs4971059 with the allele G converted to allele A, thereby resulting in TRIM46 overexpression, boosting breast carcinoma cell growth, enhancing chemotherapy resistance in vitro, and hastening tumor development in vivo [37]. In addition, Yang and colleagues (Figure 2b) reported the noncoding regulatory variant rs11836367 at the NTN4 locus (12q22) and identified it to be associated with the risk of breast carcinoma as a causal variant. The rs11837367 protective T allele promotes GATA3 binding to the distal enhancer and increases NTN4 expression [38].
2.2.2. Prostate Cancer
越来越多的证据表明,非编码区的 S
everalNP 是人类基因组中最常见的遗传变异类型,占个体间变异的 studies have90% [ independently6 identified, several57 genes]。根据位置的不同,该区域可以包含近端(启动子、增强子或超增强子)或远端(基因间或基因内)的反应元件。GWAS鉴定的风险位点位于细胞类型特异性活性染色质的基因组区域,其中大部分是数量性状位点、甲基化数量性状位点和转录因子(TF)结合相关位点。染色质构象研究有助于将 in specific prostateSNP 定位的调控区域与其各自的靶基因联系起来 [ cancer6 (PCa), susceptibility58 loci, that59]. are这些位点可能参与基因转录、转录后加工、翻译、翻译后修饰和其他调节基因表达的过程。已经使用表达数量性状位点 either controlled by causative SNPs containing a(eQTL) 鉴定了许多靶基因,以检测 SNP 与基因表达之间的关系。非编码 SNP 可以通过序列近端 ( cis-regulatory element (CRE) or have) 或远端 ( beentrans ) 调节靶基因的转录)-相互作用。研究发现,此类风险 indicated as SNP
-associated genes [39]. 区域的组蛋白修饰特别丰富,尤其是与启动子和增强子活性相关的区域(H3K4me3、H3K4me1、H3K27ac)。预计大多数 SNP
会破坏特定转录因子的结合基序。例如,rs
339331 at 6q22 was found6983267 可能会改变 MYC、CTCF 和 TCF7L2 等转录因子的结合 [ to16 be]。除了通过改变转录因子结合位点 a prostate cancer risk-associated(TFBS) 影响基因转录水平外,非编码 SNP 还会改变表观遗传修饰和/或染色质结构以影响靶基因表达。通过上述方法,非编码SNP参与细胞增殖、凋亡、迁移和侵袭。
2.1. 改变启动子的遗传变异
启动子是一段 variant.DNA The risk allele T of序列,它被识别、结合并用于启动 RNA 聚合酶的转录。启动子包含 RNA 聚合酶特异性结合和转录起始所需的保守序列的变异。大多数启动子位于结构基因转录起始点的上游,启动子本身不被转录[ rs33933160 has]。启动子位于给定结构基因 been found to augment the enhancer-binding5' 端的上游,它们激活 RNA 聚合酶以准确结合模板 DNA,特异性地诱导转录启动 [ of60 ]]. HOXB13,启动子本身不控制基因活动;相反,基因活性是通过与称为转录因子 alter the level of the RFX6 protein in an allele-specific(TF) 的蛋白质结合来调节的。启动子区域内的 SNP 通常通过影响此类转录因子的结合发挥调节作用。最近报道的一个例子是位于死亡受体 4 (DR4) 启动子的 SNP rs13278062,它会改变结直肠癌的风险。研究表明,rs13278062 G>T 变异改变了转录因子 Sp1/NF1 的结合亲和力,增加了 DR4 的表达,从而抑制了结直肠癌的发生和转移 [ manner,61 and]。MPO confer a predisposition to prostate cancer启动子 SNP rs2333227 通过改变启动子对 AP-2α 的亲和力来增加结直肠癌的恶性特征 [ [40]62].
Recently,位于 Huang et al. also identified thatMSMB 基因上游启动子的变异 SNP rs10993994 也被发现在前列腺癌患者中过度表达;这归因于更强的 CREB 结合,从而增加了启动子活性 [ the63 ]。此外,SNP
Ca-associated rs11672691
located within an enhancer element can change the binding site of HOXA2, which in turn promotes oncogenesis by impacting the expression of nearby genes [41].
Notably是与前列腺癌相关的风险基因座,与 lncRNA PCAT19 相关。非风险变体 rs11672691 及其连锁不平衡 (LD) SNP rs887391 更有可能将转录因子 NKX3.1 和 YY1 与 PCAT19-short 启动子结合,从而导致启动子增加但增强子活性降低,然后激活 PCAT19-short ,
there并最终导致较低的前列腺癌易感性 are[ other64]. casesGWAS of SNPs causing DNA-binding polymorphisms in在乳腺癌中发现了多个基因启动子区域的 SNP,包括 TERT、KLHDC7A、PIDD1 和 ESR1,报告者研究表明独立的风险等位基因会改变目标启动子活性[ distinct65、66 transcription]。大多数报告的启动子变化通过改变 factors. TF
or example, a gastric cancer risk- 结合发挥其调节作用。SNP rs3824662 等位基因 A(图2a
ssociated polymorphism (rs2978980 T>G) that is situated)通过改变 TF GATA3 表达增加染色质可及性,促进 GATA3 与 CRLF 启动子的结合,并最终形成染色质环 [ in67 an]。
图 2.非编码 SNP 使用的作用机制示意图。( a ) SNP rs3824662 等位基因 A 通过诱导 GATA3 表达、促进 GATA3 与 CRLF 启动子的结合并最终形成染色质环来增加染色质可及性。( b ) NTN4 增强子风险变体 rs11836367 与 TF GATA3 结合以调节 NTN4 表达,最终促进乳腺癌的发生和发展。( c ) 增强子 SNP rs7959129 风险等位基因 G 与启动子 SNP rs6192603 风险等位基因 G 相互作用,通过结合 TF GATA3 和 SP1 促进 ATF1 表达。( d) 风险等位基因 rs11986220 和 –10 Kb 处更高的甲基化协同作用赋予更高的肿瘤风险;然而,当-20 Kb 被低甲基化时,风险 SNP 的功能被 CTCF 介导的增强子阻断绝缘子环抑制。( e ) LINC00673 中的风险变异 rs11655237 创建了一个 miR-1231 结合位点,该位点会干扰 LINC00673 的表达并导致胰腺癌易感性。
2.2. 改变增强子的遗传变异
增强子是 intronicDNA enhancer序列的区域,可以增加其靶基因序列的顺式作用转录。每个增强子与其目标启动子的距离不同;在哺乳动物物种中,增强子可以距其目标基因 of100 lncPSCAbp has到 been foundMb [ to68]. disrupt与启动子不同,增强子可以在基因的任何地方找到;它们可以位于目标基因的上游或下游,甚至位于另一个基因的基因体内,增强子调控可以绕过其他基因,而不管它们的方向如何。增强子必须与特定的蛋白质因子结合以增强其目标的转录。增强子通常具有组织或细胞特异性,因此它们仅在某些细胞或组织中表现出活性,这取决于这些细胞或组织中存在的特定蛋白质因子 the[ binding69 of]。增强子通常由表观遗传标记 the transcription factorH3K4me1 和 H3K27ac 识别,它们存在于活性增强子元件中。相反,H3K27me3 被认为是与较低增强子活性相关的沉默表观遗传标记 [ RORA70,
thereby71 resulting]。GWAS in lower确定的常见疾病风险位点通常出现在非编码区域,其中许多被认为起到增强剂的作用 [ lncPSCA72 expression]。根据新出现的数据,这些 in an allele-specificSNP 可能通过改变重要转录因子与关键转录增强子的结合来影响基因调控 [ manner73 [42].]。
2.2.1. 乳腺癌
在所有癌症中,迄今为止发现的风险位点数量最多的是乳腺癌 As[ another13 example,]。了解恶性转化的驱动机制提供了对抗癌症复发和治疗耐药性的前景。张等。确定 the rs2647046 enhancer has been found to interact with theSNP rs4971059 位于第六个内含子和 TRIM46 基因的活性增强子元件内。通过使用 CRISPR/Cas9 介导的同源重组,他们构建了等位基因 G 转换为等位基因 A 的 SNP HLA-DQB1-AS1 promoter
to alter its
expression via a CTCF-mediated long-range loop4971059,从而导致 TRIM46 过表达,促进乳腺癌细胞生长,增强体外化疗耐药性,并加速体内肿瘤发展 [ in74 an] allele-specific manner, thereby conferring suscepti]. 此外,Yang 及其同事(图 2b
ility) to hepatocellular cancer (HCC报告了 NTN4 位点 (12q22)
[43].的非编码调控变异 Another
variation on chromosome 11q13.3 in a distants11836367,并将其确定为与乳腺癌风险相关的因果变异。rs11837367 保护性 T 等位基因促进 GATA3 与远端增强子结合并增加 NTN4 表达 [ intergenic75 region]。
2.2.2. 前列腺癌
几项研究已经独立鉴定了特定前列腺癌 h(PCa
s) 易感基因座中的几个基因,这些基因要么由含有顺式调控元件 been(CRE) characterized as a susceptibility locus for的致病 SNP 控制,要么被指定为 SNP 相关基因 [ renal76 cell]。6q22 cancer. To control transcription, the 11q13.3 locus encodes的 SNP rs339331 被发现是一种前列腺癌风险相关变异。已发现 rs339331 的风险等位基因 T 可增强 HOXB13 的增强子结合,以等位基因特异性方式改变 RFX6 蛋白的水平,并赋予前列腺癌易感性 [ a77 long-r]。最近,Huang
e enhancer that physically connects with the 等人。还发现位于增强子元件内的 PCa 相关 rs11672691 可以改变 HOXA2 CCDN1的结合位点,从而通过影响附近基因的表达促进肿瘤发生 promoter[78 [44].]。
值得注意的是,还有其他 Interestingly, SNP
sites can act案例导致不同转录因子中的 DNA 结合多态性。例如,已发现位于lncPSCA内含子增强子中的胃癌风险相关多态性 a(rs
promoters and enhancers simultaneously,2978980 T>G) 会破坏转录因子 RORA 的结合,从而以等位基因特异性方式导致较低的lncPSCA表达[ and79 their]。作为另一个例子,已发现 conversionrs2647046 增强子与HLA-DQB1-AS1启动子相互作用,以等位基因特异性方式通过 isCTCF determined by the background介导的长程环改变其表达,从而赋予对肝细胞癌 (HCC) 的易感性 [ genotype80].
As远处基因间区域染色体 a result,11q13.3 的另一个变异已被表征为肾细胞癌的易感位点。为了控制转录,11q13.3 基因座编码了一个与CCDN1启动子物理连接的远程增强子 one[ gene81 can]。有趣的是,SNP produce several different RNAs that are involved in the development of diseases. The 位点可以同时充当启动子和增强子,它们的转换由背景基因型决定。因此,一个基因可以产生几种不同的 RNA,这些 RNA 与疾病的发展有关。SNP rs11672691
mediates promoter and enhancer switching under different genotypes. A risk-associated sequence in the 介导不同基因型下的启动子和增强子转换。PCAT19-long enhancer interacts with the 长增强子中的风险相关序列与PCAT19相互作用-long
promoter to启动子通过激活细胞周期基因来促进前列腺癌的发展 [ enhance64 prostate]。
2.2.3. 大肠癌
GWAS cancer development thro已经确定了许多结直肠癌风险基因座,但只有这些基因座的一小部分靶基因被系统地询问过。例如,Yu
gh activating cell cycle genes [26].
2.2.3. Colorectal Cancer
GWAS have identified numerous colorectal cancer risk loci, but only a fraction of the target genes of these loci have been systematically interrogated. For example, Yu et al. identified a common SNP (rs7198799) in the intron of the gene
等人。在基因CDH1. They demonstrated that the risk allele C of rs7198799 acts as an enhancer that can target the TF NFATC2 and remotely enhance ZFP90 expression [45]. A prominent mechanism by which SNP variants can affect cell-specific enhancer function is via altered TF binding, thus regulating the target gene’s expression. Tian et al. identified two risk SNPs (rs61926301 and rs79591129) located in the ATF1 promoter and first intron, respectively. These are enriched in enhancer regions and open chromatin, which are also associated with H3K4me1, H3K27ac, and ATAC-seq peaks. The two variants increase the expression of ATF1 through preferentially binding to the two TFs SP1 and GATA3 [46]. Rs174575 can act as a specific remote enhancer of FADS2 and lncRNA-AP002754.2 with the participation of the transcription factor E2F1. Interestingly, TF E2F1 can promote the expression of FADS2, form a chromatin loop, and affect the occurrence of colorectal cancer [47].
2.3. Genetic Variants That Affect Promoter–Enhancer Interactions
Promoter–enhancer interactions (PEIs) underlie differential transcriptional regulation. Several technologies (chromosome conformation capture (3C), Hi-c, and H3K27Ac-HiCHIP) allow for the study of long-range cis-regulation [48][49][50]. Promoter–enhancer interactions are essential events involved in the current theory of transcriptional control. So far, there is little evidence that PEIs are required for the transcriptional control of an enhancer’s target gene. The insertion or deletion of promoters, the absence of certain PEI-associated proteins, and the inclusion of PEI-disrupting insulators all have an effect on the expression of target genes. Tian et al. found two risk variants (rs1926301 and rs7959129) located in the ATF1 promoter and intron, respectively; the former binds the TF SP1 while the latter binds the TF GATA3 (Figure 2c). They found that these two risk sites increase the interaction between the promoter and enhancer by binding SP1 and GATA3, facilitating ATF1 expression, and conferring hereditary susceptibility to CRC [46]. Moreover, the SNP rs11672691 mediates promoter and enhancer switching in a manner dependent on different background genotypes. The risk is determined by the PCAT19-long enhancer interacting with the PCAT19-long promoter, thereby altering prostate cancer development through activating cell cycle genes [26].
2.4. Genetic Variants That Alter 3D Genome Architecture
Within的内含子中鉴定了一个常见的 the nucleus, genomic DSN
A folds into a three-dimensional structure organized at different levels by the formationP (rs7198799) 。他们证明 rs7198799 的风险等位基因 C 作为增强子可以靶向 TF NFATC2 并远程增强 ZFP90 表达 [ of82]. chromatinSNP rings. These structures can bring distant enhancers near their target promoters to affect gene expression and regulation. The chromosomes fold into chromatin characterized by sequence-regulating变体影响细胞特异性增强子功能的一个重要机制是通过改变 TF 结合,从而调节靶基因的表达。田等。确定了分别位于 ATF1 启动子和第一个内含子中的两个风险 SNP(rs61926301 和 rs79591129)。这些富含增强子区域和开放染色质,它们也与 H3K4me1、H3K27ac 和 ATAC-seq 峰相关。这两种变体通过优先结合两个 TF SP1 和 GATA3 来增加 ATF1 的表达 [ spatial83 interaction]。Rs
that are key174575 可以作为 FADS2 和lncRNA-AP002754.2的特异性远程增强子在转录因子 toE2F1 maintaining normal的参与下。有趣的是,TF E2F1可促进FADS2的表达,形成染色质环,影响结直肠癌的发生[ cell84 status]。
2.3. 影响启动子-增强子相互作用的遗传变异
启动子-增强子相互作用 and function. (PEI
n cancer) 是差异转录调控的基础。几种技术(染色体构象捕获 (3C)、Hi-c 和H3K27Ac - HiCHIP)允许研究远程顺式调节[ genomes,85、86、87 structural] variation typically results in changes to the genome’s 3D structure and, as a result,。启动子-增强子相互作用是当前转录控制理论中涉及的基本事件。到目前为止,几乎没有证据表明增强子靶基因的转录控制需要 PEI。启动子的插入或缺失、某些 PEI 相关蛋白的缺失以及 PEI 破坏绝缘子的包含都会对靶基因的表达产生影响。田等。发现两个风险变体(rs1926301 和 rs7959129)位于ATF1启动子和内含子,分别;前者结合 alterationsTF in genome-mediated transSP1,而后者结合 TF GATA3(图2c
riptional)。他们发现这两个风险位点通过结合 controlSP1 [51].和 ChangesGATA3、促进 in the three-dimensional genome architectureATF1 表达并赋予 CRC 遗传易感性来增加启动子和增强子之间的相互作用 [ or83 high-order]。此外,SNP chromatin structure are linked to the development and progression of several diseases [52][53]. Longrs11672691 以依赖于不同背景基因型的方式介导启动子和增强子转换。风险由 PCAT19-long 增强子与 PCAT19-
distance chromatin lo
oping regulates cancerng 启动子相互作用决定,从而通过激活细胞周期基因改变前列腺癌的发展 [ susceptibility64 genes]。
2.4. 改变 3D 基因组结构的遗传变异
在细胞核内,基因组 eitherDNA actively or passively. Enhancers折叠成一个三维结构,通过形成染色质环在不同层次组织起来。这些结构可以将远距离增强子带到它们的目标启动子附近,从而影响基因表达和调控。染色体折叠成染色质,其特征是序列调节空间相互作用,这是维持正常细胞状态和功能的关键。在癌症基因组中,结构变异通常会导致基因组 3D 结构发生变化,从而导致基因组介导的转录控制发生改变 [ frequently88 form]。三维基因组结构或高级染色质结构的变化与多种疾病的发生和发展有关 long-range[ chromatin89 loops, with their target gene promoter regions to affect gene expression90].
The 长距离染色质环主动或被动调节癌症易感基因。增强子经常与其靶基因启动子区域形成远程染色质环以影响基因表达。例如,9q22
locus, for example, contains the thyroid cancer risk-related 基因座包含与甲状腺癌风险相关的 SNP rs965513
, which demarcates a ,它划定了与 PTC 风险密切相关的 33-kb
linkage disequilibrium block (including the lead 连锁不平衡块(包括先导 SNP rs965513
) that is strongly linked with PTC risk. The chromatin characteristics and regulatory element signatures of this block indicate at least three regulatory elements that operate as enhancers. Using chromosomal conformation capture technology, researchers have observed the long-range looping connections of these elements with the promoter region shared by )。该块的染色质特征和调控元件特征表明至少三个调控元件作为增强子起作用。使用染色体构象捕获技术,研究人员在人类乳头状甲状腺癌细胞系 (KTC-1) 和未受影响的甲状腺组织中观察到这些元件与 FOXE1
and和 PTCSC2
in a共有的启动子区域的长程环状连接 [91 human]。同样,张等人。发现 papillary thyroid cancer cell line (KTC-1) and unaffected thyroid tissue [54]. Similarly, Zhang et al. discovered that the rsrs1859962
risk-associated风险相关 LD
block contains a PCa-specific enhancer块包含一个 PCa 特异性增强子,它与SOX9基因形成一个 that forms a 1-Mb
chromatin loop with the SOX9 gene. This 染色质环。这项研究发现,rs
tudy found that the rs1859962 PCa
risk LD block contacts风险 LD 阻断通过连接到 E1 增强子的长距离染色质环接触SOX9 [ 92 via a long-distance chromatin loop that connects it to the E1 enhancer [55].
]。
CTCF
is是一种转录因子,可通过环化促进远程染色体接触。霍夫曼等人。发现 a transcription factor that promotes long-range chromosomal contact via looping. Hoffman et al. discovered that one allele in the 7 号染色体上 Igf2/H19
imprinting control region 印记控制区 (ICR)
on chromosome 7 colocalized with one allele of 中的一个等位基因与 11 号染色体上 Wsb1/Nf1
on chromosome 11. The lack of CTCF or the ablation of the maternal ICR was found to eliminate this connection and alter the expression of the 的一个等位基因共定位。CTCF 的缺乏或母体 ICR 的消融被发现消除了这种联系并改变了Wsb1/Nf1基因的表达[ gene93 [56].]。这一发现证实了 This finding confirmed the importance of CTCF
in the control of the shape of chromatin and the resulting gene expression. On the other hand, the unique contribution of CTCF is that在控制染色质形状和由此产生的基因表达方面的重要性。另一方面,CTCF 的独特贡献在于绝缘体。绝缘子是短的核苷酸序列,它决定了彼此靠近的基因组区域的边界 [ of an insulator94 ]].
Insulators当 are short nucleotide sequences that determine the boundaries of genomic areas that are close to one another [57]. When CTCF
binds与绝缘体区域结合时,它会通过干扰增强子和基因启动子之间的通讯来抑制基因转录 to[ an95 insulator]。艾哈迈德 region, it inhibits gene transcription by interfering with the communication between an enhancer and a gene promoter [58]. Ahmed M.
et等人。通过执行 al. identified (Figure 2CRISPRi 筛选识别(图2d
))非编码顺式调控元件 noncoding cis-regulatory elements (rCRE)
by。他们发现 performing CRISPRi screens. They discovered that the 8q24.21
area is widely marked with 区域广泛标记有 H3K27ac
and has a significant binding affinity to AR, FOXA1, and,并且与 AR、FOXA1 和 HOXB13
, all of which are important transcription regulators for PCa pathogenesis 具有显着的结合亲和力,所有这些都是 PCa 发病机制的重要转录调节因子 [59].[ Using96 an]。使用涉及 integrated approach involving ChIP, Hi-C, ChIP、Hi-C、CRISPR
, and functional rescue, researchers also discovered that the r 和功能拯救的综合方法,研究人员还发现包含 rCRE 序列的 rs11986220
containing the rCRE sequence interacts with the 与MYC相互作用启动子在 promoter in V16A
cells but not in 细胞中而不是在 22Rv1
cells, as the promoter–CRE interaction is typically facilitated by a细胞中,因为启动子与 CRE 的相互作用通常由上游 10 kb 区域的 CTCF
site in位点促进,从而防止染色质环化 [ a96 10 kb ]。同样,r
egion upstream, which prevents chromatin looping [59]. Similarly, the rss6702619
region is区域居住着 inhabited by CTCF, which acts as an insulator with long-range physical interactions with CRC-relevant lociCTCF,它充当绝缘体,与 CRC 相关位点进行远程物理相互作用 [ [60].97 Understanding]。了解 CTCF
-mediated 3D genomic architecture will aid in understanding the mechanism of action underlying noncoding 介导的 3D 基因组结构将有助于了解 CTCF 位点或调控增强子位点的非编码 GWAS SNP
s at either 的潜在作用机制 [ CTCF98 sites or regulatory enhancer sites [61].
]。2.5. Genetic Variants That Influence the Binding of miRNA
2.5. 影响 miRNA 结合的遗传变异
Micro微小 RNA
s (miRNA
s) are noncoding RNA molecules that influence gene expression via regulating messenger RNA degradation and translation. ) 是非编码 RNA 分子,通过调节信使 RNA 降解和翻译影响基因表达。MicroRNA
s are normally excised by the 通常被 RNase iii
enzyme酶 Dicer
from 60–110 nucleotide long hairpin precursor (folded) RNA structures (从 60-110 个核苷酸长的发夹前体(折叠)RNA 结构(pre-miRNA
s), which are then integrated into the RNA-induced silencing complex (RISC). The )中切除,然后整合到 RNA 诱导的沉默复合物(RISC)中。pro-miRNA
sequence is transcribed by 序列由 Pol-II
[62].[ Accumulating99 evidence] suggests that miRNAs play a key role in carcinogenesis by binding to the 3’-UTR of target转录。越来越多的证据表明,miRNA 通过与目标 mRNA 的 3'-UTR 结合,在致癌作用中发挥关键作用 [ mRNAs100 [63]. ]。MiRNA
mutations突变或其错误表达与人类恶性肿瘤和癌症相关基因表达的改变有关 or[ their101 misexpression have been associated with human malignancies and alterations in cancer-associated gene expression [64]. Hoffman et al. detected a variant (rs11614913) in ]。霍夫曼等人。在has-miR-196a-2中检测到一个变体 using(rs11614913)使用 GWAS
to screen genetic variants in 15 miRNAs. This SNP was identified筛选 15 种 miRNA 中的遗传变异。该 SNP 被确定与降低的乳腺癌风险相关 [ to102 be associated with decreased breast cancer risk [65]. Previous research has confir]。先前的研究已经证实,m
ed that the methylati
onRNA区域[ of103 [66] islands in ]岛的甲基化可能改变miRNA
regions 的功能,从而影响致癌途径。作者和他的同事发现,m
ay change miRNA function, thereby influencing carcinogenic pathways. The author and his colleagues found that a CpG island iniRNA 前体上游区域的 CpG 岛与乳腺癌风险相关 [ the102 region upstream of the miRN]。A
precursor is associated with breast cancer risk [65]. T
he ATF1 rs11169571 variant was shown to be strongly related to ATF1
expression by influencing rs11169571变体通过影响hsa-miR-1283 and 和hsa-miR-520d-5p结合显示与 binding, which mayATF1 表达密切相关,这可能会增加结直肠癌的易感性 [ increase susceptibility to colorectal cancer [16]56 ]].
In此外,位于 addition, SNPs located in the 3’MDM4、CD44、LAMC1 和其他基因的3'UTR
区域的SNP发挥类似的机制[ region104、105、106 of]。
长链非编码 MDM4,RNA CD44, LAMC1, and other genes exert a中的一些 similar mechanism [67][68][69].
S
ome SNP
s within long non-coding RNA can also change their binding affinity to miRNAs. The variant loci rs1317082, discovered at exon 1 of lncRNA 也可以改变它们与 miRNA 的结合亲和力。在 lncRNA RP11-362K14.5 (CCSlnc362)
,的外显子 establishes a binding site for 1 处发现的变异基因座 rs1317082为miR-4658,建立了一个结合位点,从而降低了 which consequently reduces CCSlnc362
expression and confers lowered susceptibility to CRC的表达并降低了对 CRC 的易感性 [70].[ The107 ]。l
ink between rs140618127 in the lncRNA LOCncRNA LOC146880 中的rs1406880 with non-small cell lung cancer involves a 18127与非小细胞肺癌之间的联系涉及miR-539-5p binding site. The combination of 结合位点。已发现miR-539-5p and 和LOC146880 has been found to result in the reduced activation of the oncogene 的组合导致致癌基因ENO1. Reduced 的激活减少。减少ENO1磷酸化还导致较低的 phosphorylation also results in lower PI3K and Akt activation, whichPI3K 和 Akt 激活,这与细胞增殖和肿瘤形成减少有关 [ is108 linked]。此外,LINC00673 to decreased cell proliferation and tumor formation [71]. Moreover, the 外显子中的 SNP rs11655237
allele等位基因 G
in LINC00673 exon can create a 可以创建一个 miRNA
binding site that increases the function of 结合位点,从而增加 LINC00667
表达的功能(图2e
xpression)。此外,RCC [ 109 (Figure 2e).] Furthermore,中的 rs67311347
、CRC [ in110 RCC] [72],中的 rs12982687
in和颈部鳞状细胞癌 CRC(HNSCC) [73],中的 and rs16854802
in[ neck111 squamous] cell carcinoma (HNSCC) [74] are SNPs in 是 lncRNA
sequences序列中的 that affect target gene expression by binding with miRNA. If a SNP occurs within miRNA, it will consequently affect the binding affinity of the SNP,它们通过与 miRNA 结合影响靶基因表达。如果一个 SNP 出现在 miRNA 中,它会因此影响 miRNA
to target genes.
与靶基因的结合亲和力。