Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Annamaria Lia and Version 2 by Vivi Li.
Alzheimer’s disease (AD) is a hereditary and sporadic neurodegenerative illness defined by the gradual and cumulative loss of neurons in specific brain areas. The processes that cause AD are still under investigation and there are no available therapies to halt it. PCurrent progress puts at the forefront the “calcium (Ca2+) hypothesis” as a key AD pathogenic pathway, impacting neuronal, astrocyte and microglial function. An increasing body of evidence points out the early and crucial role of cellular Ca2+ handling dysregulation in AD pathogenesis. Interestingly, Ca2+ is a key regulator of several mitochondrial functions, such as ATP production, and brain cells rely mostly on OXPHOS to match their energy demands.
- calcium
- Alzheimer’s disease
- mitochondria
- bioenergetics
- neuron
- microglia
- astrocyte
- AD mouse model
- iPSCs
1. Introduction
Mitochondria are well-known organelles that function as the powerhouse of the cell. They occupy approximately 20% of the cell volume, although their number and size vary from cell to cell. Their presence is massive in excitable cells, which rely strongly on oxidative phosphorylation (OXPHOS) to produce the adenosine triphosphate (ATP) necessary to accomplish their physiological functions [1]. However, mitochondria are much more than mere producers of energy. Decades of research have shown that mitochondria represent a formidable intracellular signaling platform that orchestrates various cellular functions. They are crucial, for example, in calcium (Ca2+) handling, regulation of cell response to stress, autophagy and cell death. In these processes, their tight interaction with other organelles, in particular the endoplasmic reticulum (ER), is fundamental [2][3][2,3]. The central role of mitochondria in cellular physiology is such that alterations in their functionality are involved in the pathogenesis of various disorders, including Alzheimer’s disease (AD).
AD is the most common neurodegenerative disease caused by poorly known pathogenic mechanisms, affecting around 10% of the population aged more than 65 in the United States [4] and around 5% of the European population [5]. Although the sporadic form (SAD) is the most prevalent, the presence of mutations in the amyloid precursor protein (APP), presenilin-1 (PSEN1) and presenilin-2 (PSEN2) genes are marks of familial cases (FAD). FAD forms represent a small percentage (2–3%) of AD cases, and PSEN1 and PSEN2 mutations account for more than 80% of genetic lesions [6]. FAD is characterized by an early onset and a worse prognosis compared to SAD, despite that they share many clinical manifestations and pathological hallmarks, i.e., accumulation and deposition of brain extracellular amyloid beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs), made by aggregates of hyper-phosphorylated tau. Besides memory and cognitive dysfunctions, behavioral and psychological symptoms of dementia (BPSD) are emerging as key clinical manifestations of AD. The severity of BPSD is shared in FAD and SAD, although their onset differs between the two groups, with a prevalence of BPSD higher in FAD compared to SAD [7]. In an attempt to clarify the pathogenic mechanisms leading to the development of AD, FAD-linked mutations have been exploited for the development of animal models that are useful for preclinical studies [8].
Over time, several hypotheses have been proposed to explain the pathogenesis of AD. The first and the most studied ones focus on the accumulation and deposition of Aβ plaques (amyloid cascade hypothesis [9]) and NFTs (tau hypothesis [10]). Despite immeasurable investigations performed in the last few decades, there are still no efficient disease-modifying treatments for AD [11]. Indeed, many clinical trials targeting Aβ and tau have failed, and the disease still lacks a cure [12]. More recently, other signaling pathways have also been investigated as pathogenic in AD, leading to the formulation of the infectious hypothesis, the inflammation hypothesis, the cholinergic hypothesis, the glutamatergic hypothesis, and many others (summarized here [13][14][13,14]). Various mitochondrial defects, such as decreased bioenergetics and ATP synthesis, increased ROS (reactive oxygen species) production, and altered mitochondrial transport and dynamics, have been reported to contribute to synaptic dysfunction and neuronal cell death in both SAD and FAD [15], leading to the formulation of the mitochondrial cascade hypothesis [16].
An increasing body of evidence points out the early and crucial role of cellular Ca2+ handling dysregulation in AD pathogenesis [17]. Interestingly, Ca2+ is a key regulator of several mitochondrial functions, such as ATP production, and brain cells rely mostly on OXPHOS to match their energy demands. Moreover, mitochondrial Ca2+ alterations may affect the functionality of other brain cells involved in memory formation and consolidation. Indeed, a growing number of studies have revealed that memory formation is not only a matter of neuronal interactions, but it also crucially involves glial cells [18]. As a result, the classical synaptic framework, consisting only of pre- and post-synaptic compartments, has been gradually expanded to include astrocytes [19] and microglia [20]. The involvement of these cell types in AD pathogenesis has also been confirmed by genome-wide association studies (GWAS). These studies describe SAD-associated genetic polymorphisms in genes involved in microglia, astrocytes and neuronal functions [21][22][23][24][21,22,23,24], as well as genes encoding mitochondria complexes or proteins involved in energy metabolism [25][26][27][28][25,26,27,28].
Thus, starting from the recent concept of quad-partite synapse and its importance in the determination of brain functions, rwesearchers will here summarize the evidence of mitochondrial Ca2+ alterations and their consequences on cell bioenergetics in AD, focusing on neurons, astrocytes and microglia. ReseaOurchers' overview will span different experimental disease models, focusing in particular on in vivo studies, but including also those based on Induced Pluripotent Stem Cells (iPSCs) that, by filling the gap due to the large evolutionary distance between mice and humans [29], represent a novel and promising tool to model human neurodegenerative diseases [30].
2. The Physiology of Brain Mitochondria: Ca2+ and Bioenergetics
Mitochondria are dynamic organelles able to change size, shape and position in a few seconds. They can move along the cytoskeleton to reach specific subcellular areas, or they undergo fission and fusion to constantly remodel their network and match local energy needs [31]. The organelles are equipped with a double membrane, the internal (IMM) and outer (OMM) mitochondrial membranes, which define the intermembrane space (IMS) and the mitochondrial matrix. The latter presents Ca2+ buffering capacity and participates in several Ca2+-mediated signaling pathways. The IMM is impermeable, even to small molecules, and hosts the electron transport chain (ETC) proteins devoted to OXPHOS. To maximize ATP synthesis, the IMM has many in-folding processes that span the mitochondrial matrix, where tricarboxylic acid (TCA, also called the Krebs cycle) occurs. The OMM is permeable to molecules up to 5 kDa because of the presence of the porin VDAC (Voltage-Dependent Anion Channel) [32]. This means that the IMS small molecule composition is similar to that of the cytosol, whereas molecules with a molecular weight > 5 kDa need specific transporters. The OMM is also the membrane involved in interactions with other organelles. Among them, the ER is one of the most important and its domains closely interacting with the OMM are called Mitochondria-Associated Membranes (MAMs). MAMs regulate numerous cellular processes, such as ER-mitochondria Ca2+ shuttling, lipid synthesis, inflammatory response, autophagy and apoptosis. These highly specialized subcellular domains are emerging as powerful signaling platforms and have been the subject of extensive studies in recent years [33]. Of note, these domains of close apposition between the ER and mitochondria result in alterations in several neurodegenerative diseases, including AD (see below and Figure 1). Although the molecular identity of proteins involved in organelle tethering is still elusive, a cell-specific molecular composition of MAMs is emerging. Recently, a neuronal-specific protein, PDZD8, has been shown to be essential for maintaining ER-mitochondria juxtaposition and neuronal Ca2+ homeostasis [34]. Moreover, Ooi et al. showed that in stress-induced hypertension rats, activation of the Sigma-1 receptor, a chaperone that localizes at MAMs [35], reduces microglia M1 polarization and neuroinflammation through MAM and mitochondrial activity modulation [36]. In astrocytes, MAMs are enriched at the endfeet, i.e., in the astrocyte processes in close contact with vessels [37], but no astrocytic-specific MAM proteins are known nowadays.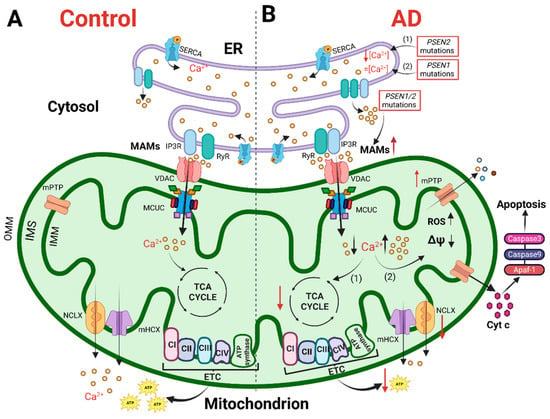
Figure 1. The cartoon shows mitochondrial Ca2+ handling and Ca2+-dependent bioenergetic functions in both physiological conditions (left) and AD (right). (A) In physiological conditions, the Ca2+ cross-talk between ER and mitochondria at MAMs, mediated by Inositol 1,4,5-trisphosphate receptors (IP3R) and ryanodine receptors (RyR) on the ER and VDAC and mitochondrial Ca2+ uniporter (MCU) complex (MCUC) on mitochondria, regulates mitochondrial Ca2+ entry, sustaining the TCA cycle and ATP synthesis. (B) In AD, various Ca2+ alterations have been reported. (1) Mutations in PSEN2 cause a reduction in the ER Ca2+ content, whereas (2) mutations in PSEN1 do not alter the ER Ca2+ content. Mutations in PSEN1/2, by increasing the open probability of ER Ca2+ releasing channels, cause an excessive release of Ca2+ from this store. Both a mitochondrial Ca2+ overload and blunted mitochondrial Ca2+ uptake ability have been described. The first stimulates a sustained production of ROS, the opening of mitochondrial permeability transition pore (mPTP) and the release of cytochrome c activating the apoptotic pathway. The second causes a decrease in ATP production, leading to a bioenergetic crisis. This original figure was created by the authors using “BioRender.com” (https://biorender.com, accessed on 7 November 2022).
3. Mitochondria in Alzheimer’s Disease: Ca2+ Signaling and Bioenergetics
More than 30 years ago, Khachaturian first described a Ca2+ homeostasis alteration in different AD cells [38][39][109,110]. From this original observation, the so-called “Ca2+ cascade hypothesis for AD” has been proposed [40][111], in which neurons from aged and diseased brains experience cytosolic Ca2+ overload upon depolarization [41][112]. Furthermore, mitochondrial Ca2+ alterations, as well as dysfunctional mitochondria, have been reported in various models of AD, including postmortem patient-derived samples, leading to the formulation of the “mitochondrial cascade hypothesis for AD” [42][113]. Based on this latter, excessive mitochondrial Ca2+ uptake triggers an increase in ROS production, ATP synthesis inhibition, mPTP opening, release of cytochrome c, activation of caspases and apoptosis (revised in [43][90], Figure 1B). However, contrasting data have also been produced, and this scheme is not completely accepted. Nowadays, there are still few works investigating the role of mitochondria in the pathogenesis of AD in physiologically relevant experimental samples, i.e., ex vivo and in vivo. Furthermore, one of the main issues to be addressed for both the Ca2+ and mitochondrial cascade hypotheses is to define whether Ca2+ or mitochondrial dysfunctions have a primary and causative role in AD pathogenesis. Indeed, to gain insight into new potential therapeutic targets, it is crucial to determine whether one of these defects is sufficient per se to cause AD. Since the mitochondrial ability to take up Ca2+ strongly depends on cytosolic Ca2+ dynamics, both parameters must be assessed in the same model. Indeed, many global Ca2+ alterations have been reported so far in various AD models.