Eosinophilic granulomatosis with polyangiitis (EGPA) is a systemic disorder characterized by peripheral eosinophilia, severe eosinophilic asthma, sinusitis, transient pulmonary infiltrates, and features of medium/small-vessel vasculitis. EGPA belongs to the group of anti-neutrophil cytoplasm antibody (ANCA)-associated vasculitides, although only 30 to 40% of patients display ANCA positivity, which is mainly of myeloperoxidase (MPO) specificity. Particularly, ANCA-positive patients typically show vasculitic features. Interleukin (IL)-5 has been demonstrated to play a crucial role in determining eosinophilic airway inflammation in EGPA patients. Specifically, maturation, activation, and survival of eosinophils especially depend on IL-5 availability. Therefore, blocking IL-5 biological activity may be a rewarding strategy for control of eosinophilic inflammation. Several monoclonal antibodies with the ability to interfere with the biological activity of IL-5 have been developed, namely, mepolizumab, reslizumab, and benralizumab. Here, we discuss the role of these drugs in the management of severe eosinophilic asthma in the context of EGPA and report the outcome of two EGPA patients with severe eosinophilic asthma treated at our outpatient clinic.
- eosinophilic granulomatosis with polyangiitis
- EGPA
- Churg–Strauss syndrome
- mepolizumab
- benralizumab
- interleukin-5
- IL-5
- severe eosinophilic asthma
1. Introduction
2. Therapeutic Management of EGPA
2.1. Standard Therapy
The cornerstone of therapy for EGPA remission induction is represented by glucocorticoids. Immunosuppressants are usually added to the glucocorticoid regimen, particularly in case of life-threatening organ involvement [12], which may be assessed with the aid of the five factor score (FFS), a prognostic tool proposed in 1996 [13] and revised in 2011 [14] (Table 1). FFS has been shown to be useful for survival prediction in EGPA; however, the original 1996 items have been refined in 2011 based on a better knowledge of the disease outcome [13,14][13][14]. Using the original set of items (1996), mortality at 5 years was observed to be 11.9% in the absence of any prognostic factor; with 1 of the 5 factors present, mortality increased to 25.9%, while with 2 or more of the 5 factors present (FFS = 2, see explanation in Table 1), mortality was shown to loom over 45.95% of the patients. In the 2011 update, age ≥ 65 years was also recognized as an independent predictor of poor prognosis, while visceral involvement was retained because it was still found to heavily weigh on the outcome. Conversely, ear, nose, and throat symptoms were associated with a lower relative risk of death. According to the 2011 set of criteria, mortality at 5 years was as follows: 9%, 21%, and 40% with FFS = 0, 1, and 2, respectively. Hence, the FFS may help identify patients requiring more aggressive treatment.| Original 1996 FFS | Revised 2011 FFS |
|---|---|
| Cardiac involvement Gastrointestinal disease (bleeding, perforation, infarction, or pancreatitis) Renal insufficiency (plasma creatinine concentration >1.6 mg/dL [141 mmol/L]) Proteinuria (>1 g/day) Central nervous system involvement |
Age > 65 years Cardiac insufficiency Renal insufficiency (stabilized peak creatinine 1.7 mg/dL [150 micromol/L]) Gastrointestinal involvement Absence of ENT manifestations (presence is associated with a better prognosis) |
2.2. Anti-IL-5-Targeted Therapies
Lung involvement is not included among the factors defining severe EGPA (Table 1); however, the lung is the most commonly involved organ in EGPA (≥90% of cases) [1,2,8,11][1][2][8][11]. Moderate-to-severe asthma is the main clinical manifestation and chest X-ray may show transient pulmonary infiltrates, which are due to the accumulation of eosinophils. Upper airway involvement is also common, in the form of chronic rhinosinusitis with nasal polyps [1,2,8][1][2][8]. Asthma is managed according to international guidelines [16]; however, the severity of symptoms may require prolonged courses with oral steroids, which are difficult to taper. Therefore, IL-5-targeted therapy may be implemented to spare glucocorticoid side effects. To date, therapeutic monoclonal antibodies capable to interfere with the IL-5 pathway and, consequently, on eosinophil growth, recruitment, activation, and survival, include mepolizumab, reslizumab, and benralizumab [17,18][17][18]. Mepolizumab and reslizumab act by sequestering soluble IL-5, making it not available to eosinophils, whereas benralizumab binds to the IL-5 receptor, thus blocking access to IL-5, but is also able to induce eosinophil killing by natural killer cells through antibody-dependent cell toxicity (ADCC) (Figure 31) [17,18][17][18]. All three monoclonal antibodies have been approved for treatment of moderate-to-severe eosinophilic asthma as add-on therapy in patients unsatisfactorily controlled by standard therapy [19], thus being theoretically administrable in EGPA-associated eosinophilic asthma as well.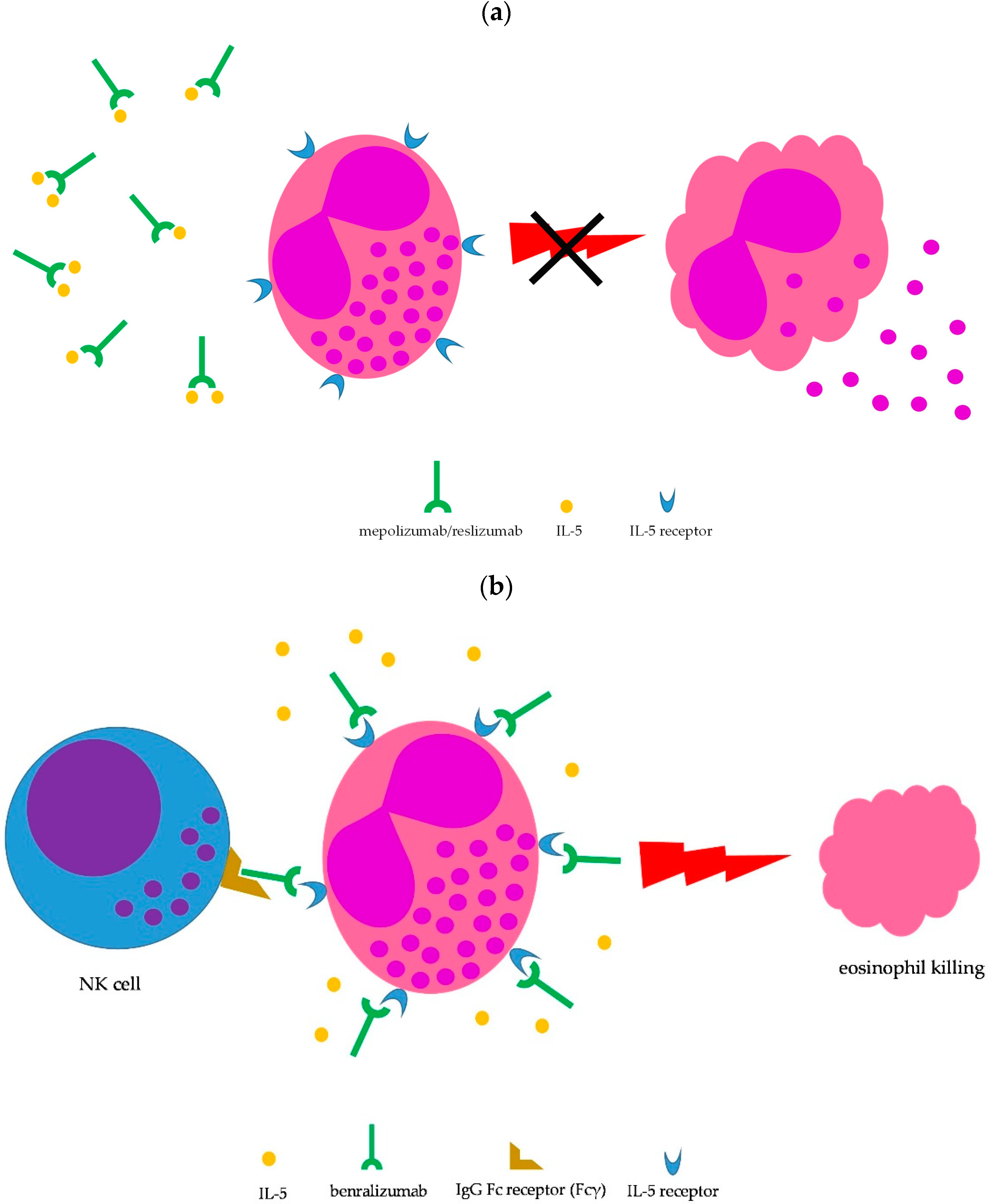
2.2.1. Mepolizumab
To date, only mepolizumab has obtained indication for relapsing/remitting or refractory EGPA [20]. In fact, following reports of encouraging results primarily in small series [21,22,23[21][22][23][24],24], a multicenter, double-blind, parallel-group, phase 3 randomized trial was eventually carried out to evaluate the efficacy and safety of mepolizumab in patients with refractory or relapsing EGPA on stable oral steroid treatment [25]. Relapses considered by the investigators included active vasculitis (Birmingham Vasculitis Activity Score [BVAS] >0 [26]), active asthma symptoms or signs, or active nasal or sinus disease, prompting an increase in the glucocorticoid dose, an initiation of or increase in immunosuppressive therapy, or hospitalization. Relapses could be of more than one type. The study confirmed the effectiveness of mepolizumab in maintaining remission throughout the study duration and in delaying major relapses when compared with placebo, with no significant between-group differences in adverse events [25]. Specifically, the study met the two primary endpoints, i.e., (1) the accrued weeks of remission (28% vs. 3%, mepolizumab vs. placebo, respectively, with the treated group showing 24+ weeks of remission) plus prednisone daily dose ≤4 mg over 52 weeks (44% successfully lowering the dose to ≤4 mg and 18% discontinuing prednisone vs. 7% and 3%, mepolizumab vs. placebo, respectively) and (2) the proportion of patients in remission at weeks 36 and 48 (32% vs. 3%, mepolizumab vs. placebo, respectively). Rates of relapses were as follows (mepolizumab vs. placebo, respectively): vasculitis 43% vs. 65%; asthma 37% vs. 60%; sinusitis 35% vs. 51%. Overall, 20% of the patients had a vasculitic relapse only, while 54% had a combined vasculitic and asthmatic/sinonasal relapse. Finally, a subgroup analysis revealed that patients with an absolute eosinophil count greater than 150 cells/mm3 at baseline were more likely to experience a greater efficacy when compared with patients with a lower value of eosinophils, who did not actually obtain a significant benefit from treatment [25]. This observation clearly paves the way for a more effective selection of EGPA patients likely responding to IL-5 therapeutic inhibition in future studies. Therefore, based on these results, the Food and Drug Administration (FDA) licensed mepolizumab in the USA in 2017 as an add-on therapy with a steroid-sparing effect in adult patients with relapsing or refractory EGPA, at a dose of 300 mg subcutaneously (s.c.) every 4 weeks. Subsequent reports from real-world clinical practice confirmed the safety and efficacy of mepolizumab in small series of patients with relapsing or refractory EGPA. Specifically, Ueno et al. [27] treated 16 patients, all of them with comorbid asthma, for one year with the licensed 300 mg dose. At the 12-month assessment, mepolizumab treatment resulted in a decreased disease activity, a remission rate of 75%, a glucorticoid-sparing effect, and a reduced number of patients using concomitant immunosuppressants. The retention rate was excellent (100% of patients). Respiratory symptoms improved immediately after starting mepolizumab, with only 2 patients still complaining of chest discomfort at the 12-month evaluation. Only three patients had an infection. The same authors also retrospectively compared the effectiveness of mepolizumab (300 mg/monthly) with i.v. cyclophosphamide pulse therapy for remission induction in EGPA patients [28]. Treatment was ≥6 months and both groups of patients were also administered high-dose steroids at the initiation of therapy. Even considering the critical issues of the study, mainly related to the small groups evaluated (7 patients in the mepolizumab group and 13 patients in the cyclophosphamide group), the results were promising: the retention rate for mepolizumab was again complete (100%), while only 61.5% of patients completed the pulse therapy (6 pulses total); adverse events were nearly twice as frequent in the cyclophosphamide group (53.8% vs. 28.6%, cyclophosphamide vs. mepolizumab, respectively). With regard to efficacy, there were no significant differences in BVAS and eosinophil count reductions between the two groups, however, mepolizumab-treated patients benefitted from a greater steroid dose reduction compared with cyclophosphamide-treated patients. Regarding lung involvement, chest manifestations decreased from 71.4% to 0% and from 58.3% to 8.3% at 6 months, mepolizumab vs. i.v. cyclophosphamide, respectively. Thus, mepolizumab for remission induction therapy in severe EGPA appeared to allow control of disease activity as effectively as cyclophosphamide, but with a greater reduction in concomitant steroid doses and fewer adverse events [28]. Finally, a small series (11 EGPA patients, 3 with uncontrolled asthma), characterized by steroid dependence and unsatisfactory disease control, also achieved improvement of disease activity (no evidence of asthma and vasculitic manifestations at the last study follow-up) following treatment with mepolizumab [29]. Again, the improvement in disease activity allowed notable glucocorticoid tapering.2.2.2. Reslizumab
Fewer evidence is available for reslizumab as a controller drug in EGPA. A study involving nine patients with refractory, steroid-dependent EGPA patients with severe eosinophilic asthma were treated with reslizumab infusions at a dose of 3 mg/kg every 28 days [37][30], according to the regimen approved as add-on therapy for treatment of severe asthma with an eosinophilic phenotype [38][31]. Patients also had other EGPA features, namely, paranasal sinus disease (100% of patients), neurological involvement (44% of patients), and cardiac involvement (33% of patients). At the end of the 48 weeks of treatment, 7 out of 9 patients (~78%) had their steroid doses reduced to or below 7.5 mg/day (consistent with remission [39][32]), while two patients were able to completely stop the drug. No increases in exacerbation frequency or hospitalization were seen and no treatment-limiting adverse effects were recorded. Since no significant improvement in overall BVAS scores was seen, the authors concluded that reslizumab may be less effective in controlling extrapulmonary manifestations of EGPA, namely, neuropathy, than in quenching activity of airway disease [37][30]. Thus, reslizumab may be a therapeutic option in EGPA patients mainly troubled by severe eosinophilic asthma. Another open-label, pilot study involving 10 patients with EGPA [40][33] demonstrated a significant reduction in the daily oral glucocorticoid dose after 7 monthly reslizumab treatments (equivalent to a 24-week treatment phase). Although only 6 of the 10 subjects completed all the study visits, 8 patients could be nonetheless analyzed; 5 out of these 8 patients were considered true responders to reslizimumab because of successful tapering of oral steroids, with no exacerbations during the treatment phase. Overall, three exacerbations were recorded during the treatment, and the most common was the worsening of asthma symptoms. One patient suffered from a severe adverse event, deemed to be causally linked with the study drug, requiring removal from the study [40][33]. Because of the small numbers of patients and the design of these studies, randomized, controlled clinical trials are needed to validate the efficacy and safety of reslizumab for EGPA.2.2.3. Benralizumab
Efficacy of benralizumab in EGPA is mainly supported by case reports [41][34]. In addition, a small open-label pilot study including 10 EGPA patients suggested that benralizumab was effective in reducing both the daily dose of oral steroids and the exacerbation rate during treatment [42][35]. Importantly, 8 of 10 subjects reached a minimum oral steroid dose of <5 mg daily, with 5 (50%) able to stop oral glucocorticoids after 40 weeks of treatment [42][35]. Intriguingly, benralizumab has been reported to be effective even in EGPA patients failing previous treatment with mepolizumab or reslizumab. In 2 case studies (16 EGPA patients overall), including 6 patients failing previous treatment with mepolizumab and 1 patient failing both mepolizumab and reslizumab, benralizumab (30 mg s.c. every 4 weeks for the first 3 injections, then 30 mg s.c. every 8 weeks) significantly reduced oral glucocorticoid doses and improved both BVAS and ACT scores at the completion of the studies [43,44][36][37]. Complete depletion of peripheral eosinophils was seen in most of the patients, consistent with the drug’s mechanism of action (Figure 31b). Currently, a phase three trial comparing benralizumab with mepolizumab in relapsing or refractory EGPA is ongoing (MANDARA trial, NCT04157348) [45][38].References
- Almaani, S.; Fussner, L.A.; Brodsky, S.; Meara, A.S.; Jayne, D. ANCA-Associated Vasculitis: An Update. J. Clin. Med. 2021, 10, 1446.
- Kitching, A.R.; Anders, H.J.; Basu, N.; Brouwer, E.; Gordon, J.; Jayne, D.R.; Kullman, J.; Lyons, P.A.; Merkel, P.A.; Savage, C.O.S.; et al. ANCA-associated vasculitis. Nat. Rev. Dis. Prim. 2020, 6, 71.
- Radice, A.; Bianchi, L.; Sinico, R.A. Anti-neutrophil cytoplasmic autoantibodies: Methodological aspects and clinical significance in systemic vasculitis. Autoimmun. Rev. 2013, 12, 487–495.
- Cornec, D.; Cornec-Le Gall, E.; Fervenza, F.C.; Specks, U. ANCA-associated vasculitis—Clinical utility of using ANCA specificity to classify patients. Nat. Rev. Rheumatol. 2016, 12, 570–579.
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 1–11.
- Prete, M.; Indiveri, F.; Perosa, F. Vasculitides: Proposal for an integrated nomenclature. Autoimmun. Rev. 2016, 15, 167–173.
- Koike, H.; Nishi, R.; Yagi, S.; Furukawa, S.; Fukami, Y.; Iijima, M.; Katsuno, M. A Review of Anti-IL-5 Therapies for Eosinophilic Granulomatosis with Polyangiitis. Adv. Ther. 2022. online ahead of print.
- Comarmond, C.; Pagnoux, C.; Khellaf, M.; Cordier, J.F.; Hamidou, M.; Viallard, J.F.; Maurier, F.; Jouneau, S.; Bienvenu, B.; Puéchal, X.; et al. French Vasculitis Study Group. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): Clinical characteristics and long-term followup of the 383 patients enrolled in the French vasculitis study group cohort. Arthritis Rheum. 2013, 65, 270–281.
- Chaigne, B.; Guillevin, L. Vasculitis for the internist: Focus on ANCA-associated vasculitis. Intern. Emerg. Med. 2017, 12, 577–585.
- Zarka, F.; Veillette, C.; Makhzoum, J.P. A Review of Primary Vasculitis Mimickers Based on the Chapel Hill Consensus Classification. Int. J. Rheumatol. 2020, 2020, 8392542.
- Grayson, P.C.; Ponte, C.; Suppiah, R.; Robson, J.C.; Craven, A.; Judge, A.; Khalid, S.; Hutchings, A.; Luqmani, R.A.; Watts, R.A.; et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Eosinophilic Granulomatosis with Polyangiitis. Ann. Rheum. Dis. 2022, 81, 309–314.
- Chung, S.A.; Langford, C.A.; Maz, M.; Abril, A.; Gorelik, M.; Guyatt, G.; Archer, A.M.; Conn, D.L.; Full, K.A.; Grayson, P.C.; et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Care Res. 2021, 73, 1088–1105.
- Guillevin, L.; Lhote, F.; Gayraud, M.; Cohen, P.; Jarrousse, B.; Lortholary, O.; Thibult, N.; Casassus, P. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine 1996, 75, 17–28.
- Guillevin, L.; Pagnoux, C.; Seror, R.; Mahr, A.; Mouthon, L.; Toumelin, P.L.; French Vasculitis Study Group (FVSG). The Five-Factor Score revisited: Assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine 2011, 90, 19–27.
- Fraenkel, L.; Bathon, J.M.; England, B.R.; St. Clair, E.W.; Arayssi, T.; Carandang, K.; Deane, K.D.; Genovese, M.; Huston, K.K.; Kerr, G.; et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res. 2021, 73, 924–939.
- Diagnosis and Management of Difficult-to-Treat & Severe Asthma—Global Initiative for Asthma—GINA (ginasthma.org). Available online: https://ginasthma.org/severeasthma/ (accessed on 23 October 2022).
- Desai, M.; Oppenheimer, J. Biologics in allergic and immunologic diseases: Promises and challenges in the era of personalized medicine. Ann. Allergy Asthma Immunol. 2018, 120, 350–353.
- van de Veen, W.; Akdis, M. The use of biologics for immune modulation in allergic disease. J. Clin. Investig. 2019, 129, 1452–1462.
- Agache, I.; Akdis, C.A.; Akdis, M.; Canonica, G.W.; Casale, T.; Chivato, T.; Corren, J.; Chu, D.K.; Del Giacco, S.; Eiwegger, T.; et al. EAACI Biologicals Guidelines—Recommendations for severe asthma. Allergy 2021, 76, 14–44.
- Nucala, INN-Mepolizumab (Europa.eu). Available online: https://www.ema.europa.eu/en/documents/product-information/nucala-epar-product-information_en.pdf (accessed on 23 October 2022).
- Kahn, J.-E.; Grandpeix-Guyodo, C.; Marroun, I.; Catherinot, E.; Mellot, F.; Roufosse, F.; Blétry, O. Sustained response to mepolizumab in refractory Churg-Strauss syndrome. J. Allergy Clin. Immunol. 2010, 125, 267–270.
- Kim, S.; Marigowda, G.; Oren, E.; Israel, E.; Wechsler, M.E. Mepolizumab as a steroid-sparing treatment option in patients with Churg-Strauss syndrome. J. Allergy Clin. Immunol. 2010, 125, 1336–1343.
- Moosig, F.; Gross, W.L.; Herrmann, K.; Bremer, J.P.; Hellmich, B. Targeting interleukin-5 in refractory and relapsing Churg-Strauss syndrome. Ann. Intern. Med. 2011, 155, 341–343.
- Herrmann, K.; Gross, W.L.; Moosig, F. Extended follow-up after stopping mepolizumab in relapsing/refractory Churg-Strauss syndrome. Clin. Exp. Rheumatol. 2012, 30 (Suppl. S70), S62–S65.
- Wechsler, M.E.; Akuthota, P.; Jayne, D.; Khoury, P.; Klion, A.; Langford, C.A.; Merkel, P.A.; Moosig, F.; Specks, U.; Cid, M.C.; et al. Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis. N. Engl. J. Med. 2017, 376, 1921–1932.
- Stone, J.H.; Hoffman, G.S.; Merkel, P.A.; Min, Y.I.; Uhlfelder, M.L.; Hellmann, D.B.; Specks, U.; Allen, N.B.; Davis, J.C.; Spiera, R.F.; et al. A disease-specific activity index for Wegener’s granulomatosis: Modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis Rheum. 2001, 44, 912–920.
- Ueno, M.; Miyagawa, I.; Nakano, K.; Iwata, S.; Hanami, K.; Fukuyo, S.; Kubo, S.; Miyazaki, Y.; Kawabe, A.; Yoshinari, H.; et al. Effectiveness and safety of mepolizumab in combination with corticosteroids in patients with eosinophilic granulomatosis with polyangiitis. Arthritis Res. Ther. 2021, 23, 86.
- Ueno, M.; Miyagawa, I.; Aritomi, T.; Kimura, K.; Iwata, S.; Hanami, K.; Fukuyo, S.; Kubo, S.; Miyazaki, Y.; Nakayamada, S.; et al. Safety and effectiveness of mepolizumab therapy in remission induction therapy for eosinophilic granulomatosis with polyangiitis: A retrospective study. Arthritis Res. Ther. 2022, 24, 159.
- Ríos-Garcés, R.; Prieto-González, S.; Hernández-Rodríguez, J.; Arismendi, E.; Alobid, I.; Penatti, A.E.; Cid, M.C.; Espígol-Frigolé, G. Response to mepolizumab according to disease manifestations in patients with eosinophilic granulomatosis with polyangiitis. Eur. J. Intern. Med. 2022, 95, 61–66.
- Kent, B.D.; d’Ancona, G.; Fernandes, M.; Green, L.; Roxas, C.; Thomson, L.; Nanzer, A.M.; Kavanagh, J.; Agarwal, S.; Jackson, D.J. Oral corticosteroid-sparing effects of reslizumab in the treatment of eosinophilic granulomatosis with polyangiitis. ERJ Open Res. 2020, 6, 00311–2019.
- Available online: https://www.cinqairhcp.com/globalassets/cinqair-hcp-redesign/prescribing-information.pdf (accessed on 23 October 2022).
- Hellmich, B.; Flossmann, O.; Gross, W.L.; Bacon, P.; Cohen-Tervaert, J.W.; Guillevin, L.; Jayne, D.; Mahr, A.; Merkel, P.A.; Raspe, H.; et al. EULAR recommendations for conducting clinical studies and/or clinical trials in systemic vasculitis: Focus on anti-neutrophil cytoplasm antibody-associated vasculitis. Ann. Rheum. Dis. 2007, 66, 605–617.
- Manka, L.A.; Guntur, V.P.; Denson, J.L.; Dunn, R.M.; Dollin, Y.T.; Strand, M.J.; Wechsler, M.E. Efficacy and safety of reslizumab in the treatment of eosinophilic granulomatosis with polyangiitis. Ann. Allergy Asthma Immunol. 2021, 126, 696–701.e1.
- Koga, Y.; Aoki-Saito, H.; Kamide, Y.; Sato, M.; Tsurumaki, H.; Yatomi, M.; Ishizuka, T.; Hisada, T. Perspectives on the Efficacy of Benralizumab for Treatment of Eosinophilic Granulomatosis with Polyangiitis. Front. Pharmacol. 2022, 13, 865318.
- Guntur, V.P.; Manka, L.A.; Denson, J.L.; Dunn, R.M.; Dollin, Y.T.; Gill, M.; Kolakowski, C.; Strand, M.J.; Wechsler, M.E. Benralizumab as a Steroid-Sparing Treatment Option in Eosinophilic Granulomatosis with Polyangiitis. J. Allergy Clin. Immunol. Pract. 2021, 9, 1186–1193.e1.
- Padoan, R.; Chieco Bianchi, F.; Marchi, M.R.; Cazzador, D.; Felicetti, M.; Emanuelli, E.; Vianello, A.; Nicolai, P.; Doria, A.; Schiavon, F. Benralizumab as a glucocorticoid-sparing treatment option for severe asthma in eosinophilic granulomatosis with polyangiitis. J. Allergy Clin. Immunol. Pract. 2020, 8, 3225–3227.e2.
- Nanzer, A.M.; Dhariwal, J.; Kavanagh, J.; Hearn, A.; Fernandes, M.; Thomson, L.; Roxas, C.; Green, L.; D’Ancona, G.; Agarwal, S.; et al. Steroid-sparing effects of benralizumab in patients with eosinophilic granulomatosis with polyangiitis. ERJ Open Res. 2020, 6, 00451–2020.
- Available online: https://clinicaltrials.gov/ct2/show/NCT04157348 (accessed on 23 October 2022).