Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Giancarlo Castaman and Version 2 by Amina Yu.
Gaucher disease (GD) is an autosomal recessive lysosomal storage disease that is caused by deficiency of the enzyme β-glucocerebrosidase (β-GCase), which is required for the degradation of glycosphingolipids. Deficiency of β-GCase is responsible for the accumulation of glucosylceramide and its deacylated product glucosylsphingosine into lysosomes of reticuloendothelial cells. These lipid-laden cells are known as Gaucher cells. Gaucher cells are large and characterized by eccentric nuclei, condensed chromatin and cytoplasm with heterogeneous “crumpled tissue paper”. The bone marrow, spleen and liver are particularly infiltrated by these cells in GD, leading to the main clinical signs of the disease at diagnosis.
- Gaucher disease
- bleeding
- hemostasis
1. Gaucher Disease (GD) and Inflammation
The multi-organ diffuse infiltration by Gaucher cells alone cannot explain the multiple and heterogeneous manifestations of the disease. The accumulation of the β-GCase substrate leads to a secondary activation of macrophages associated with an autophagy disruption and an onset of a cascade of inflammatory events that further worsen the disease [1][2][3][4][29,30,31,32].
Increased expression of tumor necrosis factor-α (TNF-α interleukin-1β (IL-1β)), interleukin-1 receptor antagonist, soluble interleukin-2 receptor (sIL-2R), interleukin-6 (IL-6), interleukin-8 (IL-8), interleukin-10 (IL-10), CD14, CD163s, MIP-1β, TGFβ, M-CSF, CCL-18, and chitotriosidase have been found in patient plasma [5][6][7][8][9][10][33,34,35,36,37,38].
The pathogenetic mechanisms of inflammatory propagation in GD have yet to be delineated, but a possible pathway has been proposed [11][39]. The primary Gaucher cells activation triggers the release of monocytes (MOs) and polymorphonuclear neutrophils (PMNs) recruiting cytokines and chemokines. Monocyte chemoattracting protein-1 (MCP-1) recruits circulating MOs, whereas IL8 and TNF-a induce the PMNs migration into the different visceral organs. With migration from the blood into the visceral organs, MOs mature into tissue-specific macrophages with GCase defect, leading to an increase in Gaucher cells and further release of IFN-g, IL-4, IL-6, and TGF-b. IFN-g and IL-4 cause the development of T helper-1 (Th1) and Th2 cell-mediated responses, whereas IL-6 induces the development of T-follicular cells, thus activating B-cells in germinal centers with associated hypergammaglobulinemia. IL-6 together with TGF-b impact Th17 cell development with IL-17 production and subsequently that of IL-8 with recruitment of circulating PMNs in GD visceral organs. In addition to IL-8, Gaucher cells also secrete IL-1b, INF-g, and TNF-a, which are crucial for recruiting PMNs with subsequent release of their activation products (TNF-a, IL-6, IL-1a, IL-1b, IL-1Ra) into the visceral organs. Moreover, TNF-a together with INF-g and IL-1b induce nitric oxide synthase (NOS) with nitric oxide (NO) production to trigger immunological inflammation in GD.
2. Inflammation and Hemostasis
Extensive cross talk exists between inflammation and coagulation, whereby inflammation leads to the activation of coagulation, and coagulation considerably affects inflammatory activity (Figure 1). Proinflammatory cytokines IL-1, IL-6, and TNF-a stimulate the production of tissue factor (TF), a transmembrane glycoprotein that serves as a surface receptor for coagulation factor (F) VIIa. The TF-FVIIa bond plays a key role for the onset of coagulation and thrombin generation [12][40]. Conversely, activated coagulation proteases may affect specific receptors on inflammatory cells and endothelial cells modulating the inflammatory response [13][41].
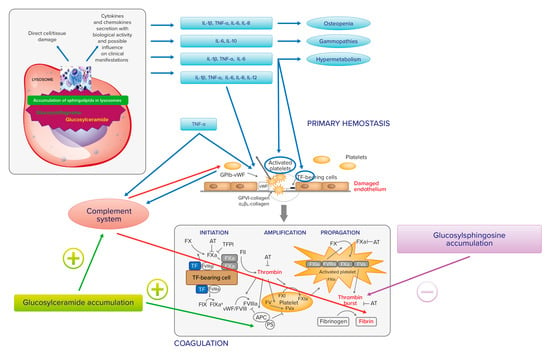
Figure 1.
Relationship between Gaucher cell accumulation, proinflammatory cytokines, and hemostasis.
IL-1, IL-6, and TNF-a may also trigger endothelial cells to switch into a procoagulant, clot-promoting state, changing their antithrombotic properties. The same cytokines, together with IL-8 and IL-12, cause platelet activation and clumping [14][15][42,43]. In addition, upregulated TNF-a may induce complement component 3 (C3). TNF-a and C3 interact with receptors on platelets. TNF-a, via tumor necrosis factor receptors 1 and 2 (TNFR1 and TNFR2), causes platelet consumption and activation through arachidonic acid pathway stimulation. C3 also interacts with the surface of activated platelets, as well as with other components of the complement system. Platelets may also interact with the complement system through non-classical complement receptor proteins, such as P-selectin or GP1ba. [16][44]. Finally, the complement plays also an additional role in fibrin deposition [17][45]. In experimental and clinical GD, a direct relationship between the accumulation of glucosylceramide and the C5a and the C5a receptor 1 axis has been observed [18][46].
In turn, activated platelets express and release several pro- and anti-inflammatory molecules, which recruit and capture circulating leukocytes and direct them to inflamed tissues. Platelets can also influence adaptive immune responses via the secretion of CD40 and CD40L molecules. Platelet membrane CD40/CD40L allows interaction with different immune cells. Platelet CD40L stimulates the release of IL-8 and MCP-1 from endothelial cells and recruits leukocytes to generate platelet–leukocyte aggregates in areas of vascular inflammation via the expression on platelet surface of adhesion molecules (E-selectin, VCAM, and ICAM-1). Moreover, platelet CD40L induces isotype switching in B-cells and enhances CD8+T-cell responses. Platelets also express CD40 and integrin aIIbb3, receptors for CD40L, by creating feedback loops from the bond.
In addition, platelet granules contain several chemokines and cytokines. CXCL4 or platelet factor 4 (PF4) together with CCL5 enhance the arrest of MOs on endothelial cells and promote their survival, chemotaxis, and differentiation. CXCL7 plays a key role in the recruitment of neutrophils. Platelet release of PF4 and sCD40L upregulates the release of co-stimulatory molecules and proinflammatory cytokines from the dendritic cells. Platelets can directly interact with dendritic cells via CD62P/CD61P, influencing their maturation and enhancing the synthesis of the Th2 helper chemokine CCL17 [19][47].
3. Glycolipids and Hemostasis
Glycolipids are important components of cell membranes and can play critical roles as bioregulators of different processes, such as blood coagulation. Different membrane phospholipid components differently affect coagulation and anticoagulation reactions. For instance, anionic phospholipidis, mainly phosphatidylserine, increase prothrombinase activity; in contrast, posphatidylethanolamine and cardiolipin enhance the activated protein C (APC) anticoagulant pathway.
Thrombin generation is inhibited by glucosylsphingosine, but not by glucosylceramide [20][48]. In turn, glucosylceramide enhances inactivation of factor Va by APC and protein S, which could represent a potential risk factor for venous thromboembolism [21][22][49,50].
It clearly appears that the links between the accumulation of glucosylceramide and glucosylsphingosine, inflammation, and hemostasis in GD may be many and different. At present, however, these are more theoretical links, of which the clinical impact on hemostasis has yet to be outlined. Figure 1 summarizes the relationship between Gaucher cell accumulation, proinflammatory cytokines, and hemostasis.
Gaucher cell accumulation represents the pathogenetic first step of Gaucher disease. On the one hand, it causes direct tissue damage by infiltration; on the other hand, it is responsible for inflammatory cascade activation, with cytokines and chemokines secretion with biological activity and influence on clinical manifestation. IL-1b, TNF-a, IL-6, and IL-10 may contribute to osteopenia; IL-6 and IL-10 may be responsible for gammopathies and multiple myeloma; IL-1b, TNF-a, and IL-6 may have a role in the activation of hypermetabolism. There is also an extensive cross talk between inflammation and hemostasis. On primary hemostasis, proinflammatory cytokines IL-1, IL-6, and TNF-a may trigger endothelial cells to change their antithrombotic properties into a procoagulant, clot-promoting state. The same cytokines, together with IL-8 and IL-12, cause platelet activation and clumping. TNF-a may also induce complement component 3 (C3) and both induce platelet consumption and activation through arachidonic acid pathway stimulation. IL-1, IL-6, and TNF-a stimulate the production of tissue factor (TF), which serves as a surface receptor for coagulation factor (F) VIIa triggering the extrinsic coagulation pathway. The TF–FVIIa bond plays a key role for the onset of coagulation and thrombin generation. The complement system, induced by TNF-a, plays a role in fibrin deposition. The accumulation of glucosylceramide and glucosylsphingosine also has a direct action on hemostasis. In fact, thrombin generation is inhibited by glucosylsphingosine and glucosylceramide can enhance the inactivation of factor Va by APC and protein S.
4. Hemostatic Management
Even though ERT and SRT may improve and correct hemostatic abnormalities in GD, treatments meant to enhance hemostasis may be necessary in specific clinical circumstances, such as recent onset of ERT/SRT, surgery, delivery, and life-threatening bleeding.
A laboratory screening assessment is required, with an evaluation of platelet count and platelet function tests, i.e., platelet function analyzer (PFA-100) [23][85] and/or platelet aggregation, APTT, and PT, in order to provide the most appropriate hemostatic treatment. When an abnormality of coagulation screening test is found, a diagnostic deepening may be recommended. After the exclusion of FVIII, FIX, and FXII defects, an isolated prolonged APTT suggests FXI deficiency, and an isolated PT prolongation is typically caused by a FVII deficiency. The combined prolongation of APTT and PT suggests the diagnosis of combined FV + FVIII, FX, FV, FII, or fibrinogen deficiencies.
Thromboelastography (TEG) may be an alternative option to evaluate the bleeding tendency in a peri-surgical or predelivery setting [24][86]. TEG provides a quick and visualized monitoring and analysis of the viscoelastic properties of clot formation and dissolution in whole blood. In addition, the separate effects of platelets and fibrinogen on overall clot strength can be rapidly assessed [25][87]. The goal of TEG is the evaluation at the point of care for the management decision of the need for blood transfusion or fibrinogen supplementation. However, the experience of using TEG in patients with GD is still limited and the assay is not performed in a sufficiently uniform manner worldwide [26][88].
5. Hemostatic Agents
Several therapeutic options to enhance an impaired hemostatic function are available, ranging from antifibrinolytics to replacement therapy with platelet transfusion or clotting factor concentrates. Tranexamic acid represents an important therapeutic aid for the management of mucocutaneous bleeding [27][89]. This synthetic derivative of the amino acid lysine exerts its antifibrinolytic effect through the reversible blockade of lysine binding sites on plasminogen molecules. It is a cheap and easy to use drug for the treatment and prevention of bleeding. Tranexamic acid is used at a dose of 15–25 mg/kg every 8 h for 3–6 days by oral or intravenous administration for bleeding or surgery. In addition, it can be used as mouthwash for oral bleeding or dental procedures. No evidence of increased thromboembolic or other significant adverse events has been reported in different large populations treated with tranexamic acid.
To control gynecological bleeding, hormone therapy alone or in combination with tranexamic acid is often used [28][29][56,90]. Oral contraceptives containing both progestin and estrogen should be started first. In women who failed to have a clinically useful response, a levonorgestrel-releasing intrauterine device may be recommended, such as in women with inherited bleeding disorders [30][91].
When tranexamic acid alone is not able to guarantee an effective hemostasis, desmopressin may be employed. Desmopressin (DDAVP, 1-deamino-8-D-arginin-vasopressin) is a synthetic analog of the antidiuretic hormone vasopressin, V2 agonist, able to increase VWF and FVIII in plasma, and, therefore, it is the drug of choice for mild hemophilia A and type 1 von Willebrand disease [31][92]. DDAVP is also clinically efficacious in patients with other, generally mild platelet function disorders of the [32][33][34][35][36][93,94,95,96,97], and its use is recommended for bleeding prophylaxis for situations at low risk of bleeding risk [37][98]. DDAVP-induced the rise of circulating VWF high-molecular-weight multimers, leading to an increased platelet adhesion to the injured vessel wall, which could enhance hemostasis in patients with platelet function disorders and in those with GD [38][39][99,100]. In addition, in vivo DDAVP strengthens the ability to form procoagulant platelets and increases platelet-dependent thrombin generation by inducing intracellular Na+ and Ca2+ fluxes [40][101]. DDAVP can be administered intravenously (0.3 µg/kg diluted in 50–100 mL saline and infused over 15–30 min), subcutaneously at the same dose, or intranasally (fixed doses of 300 µg in adults and 150 µg in children). Subsequent doses may be administered every 12–24 h, but a progressive reduction in magnitude of the response (tachyphylaxis) is observed after closely-spaced doses. Side effects, due to the vasomotor effect of the molecule, may include mild tachycardia, flushing, and headache. Rare side effects attributable to the antidiuretic properties of DDAVP are hyponatremia, especially in children below the age of 2, and volume overload. DDAVP should be used with caution in elderly subjects with atherosclerotic disease [41][102].
In GD patients with severe thrombocytopenia and/or severe functional defects, platelet transfusion may be recommended, as well as specific clotting factor concentrates administration when a serious coagulation defect is diagnosed [42][103]. Figure 2 summarizes the therapeutic options to enhance hemostasis and their mechanisms of action.
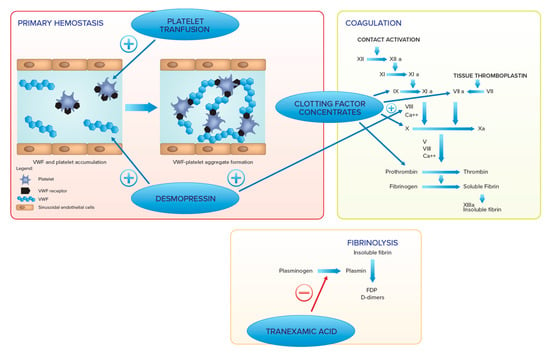
Figure 2.
Therapeutic options to enhance hemostasis.
Several agents with action on different hemostatic phases are available. In primary hemostasis, platelet transfusion increases platelet number and improves their function, whereas desmopressin increases von Willebrand factor and FVIII and improves platelet adhesion. In the coagulation pathway, desmopressin increases FVIII, whereas clotting factor concentrates increase levels of corresponding coagulation factors (e.g., FII, FVII, FIX, FX, FVIII).
In the fibrinolysis, tranexamic acid blocks plasmin generation and can be administered alone or with all the other treatments.