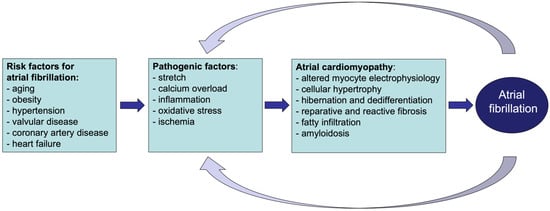
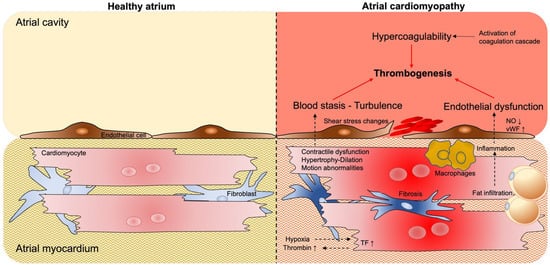
Figure 2. Atrial cardiomyopathy contributes to thrombogenesis. Unlike in the healthy atrium (
left side), in the cardiomyopathic atrium (
right side), pathological structural and functional changes (e.g., contractile dysfunction, atrial dilation, fibrosis, and fat infiltration) lead to aberrant blood flow and stasis in the atria cavity, endothelial dysfunction (and structural changes), and hypercoagulability, predisposing patients to thrombogenic events within the atrial cavity. Furthermore, hypoxic conditions, together with vascular leakage, may contribute to the activation of the coagulation cascade within the myocardial tissue. Abbreviations: NO = nitic oxide; vWF = von Willebrand factor; TF = tissue factor.
2.1. Blood Stasis and Endothelial Dysfunction
Atrial contractile remodeling leads to reduced and/or dyssynchronous atrial contraction and wall motion disturbances. This results in blood stasis, which critically contributes to thrombogenesis.
During the first days after AF onset, loss of synchronized atrial contraction goes hand in hand with electrical remodeling processes
[16][63]. Interestingly, although electrical remodeling is reversible upon sinus rhythm (SR) restoration, the impairment of atrial contractility partially remains after the cardioversion to SR, increasing the risk of thrombus formation and stroke
[17][18][19][20][64,65,66,67].
The loss of atrial contractility contributes to thrombogenesis via multiple other mechanisms. As recently demonstrated by Spartera and colleagues, a left atrial myopathic phenotype, including reduced left atrial function, is associated with altered left atrial flow characteristics in patients at moderate-to-high risk of stroke, regardless of a history of AF
[21][68]. In fact, altered atrial flow velocity and vorticity are expected to reduce endocardial shear stress. This phenomenon has been shown to downregulate the endothelial production of nitric oxide, which mediates vasodilation and has anti-thrombotic properties
[22][69]. The downregulation of atrial nitric oxide would, therefore, not only stimulate the aggregation of platelets, but also increase the expression of the protein plasminogen activator inhibitor-1 (PAI-1), resulting in impaired fibrinolysis
[14][61]. Moreover, atrial contractile dysfunction has been associated with atrial dilation, which is an independent risk factor for thrombogenesis in patients with and without AF
[23][24][25][26][25,46,70,71]. In fact, atrial dilation and volume overload of the left atrial appendage are associated with increased endocardial expression of the glycoprotein von Willebrand Factor (vWF), a well-documented marker of endothelial dysfunction
[27][28][29][72,73,74]. vWF mediates platelet adhesion to the activated endothelium, and its plasma levels are an independent predictor of poor outcome, including thromboembolic events, in patients with AF
[30][75].
The deterioration of endothelial function in atrial cardiomyopathy can also result from inflammatory processes.
As Srecently reviewed elsewhere, systemic and local (atrial) inflammation is a well-documented phenomenon in AF
[31][32][76,77]. Within the atria, inflammation leads to areas of endothelial denudation and predisposes patients to thrombotic aggregation
[33][78]. The exposure of tissue factor (TF)-expressing subendothelium to the bloodstream, as a consequence of endothelial denudation, may facilitate the activation of the coagulation cascade within the atrial cavity
[34][79]. Moreover, pro-inflammatory stimuli can directly support thrombotic events by upregulating the expression of vWF and TF in endothelial cells and monocytes
[35][36][80,81].
2.2. Pro-Thrombotic Interstitial Changes
During the complex etiology of atrial cardiomyopathy, with a variety of molecular and structural changes taking place in the atrial tissue, pro-thrombotic and pro-inflammatory changes may also be observed within the interstitial space of the atrial myocardium itself (
Figure 2). For example, the accumulation of epicardial adipose tissue (EAT) may play a role in the development of AF. Several studies have reported that EAT volume may represent an independent risk factor for AF development and a predictor of AF recurrence in patients undergoing AF ablation
[37][38][82,83]. The exact role of EAT in AF development still requires clarification. As reported by Antonopoulos and colleagues, EAT may play a protective role in the heart by decreasing myocardial oxidative stress via the secretion of adiponectin
[39][84].
Nevertheless, EAT is associated with fatty infiltration from the epicardial layer, which may cause disorganized conduction within the atria
[40][85]. Moreover, both EAT and fatty infiltration are active sources of pro-inflammatory cytokines (e.g., monocyte chemoattractant protein-1 (MCP-1), Interleukin-6 (IL-6), and tumor necrotic factor-alpha (TNF-α), which can aggravate the effect of existing pro-inflammatory processes on the endocardial endothelium and support the infiltration of immune cells within the myocardium
[7][8].
2.3. Hypercoagulability
Another mechanism that contributes to thrombogenesis in AF and in other atrial cardiomyopathies consists of alterations in blood constituents which confer a hypercoagulable state
[41][93].
Hypercoagulability in AF patients is often reflected by increased systemic platelet activation, elevated concentrations of pro-thrombotic indices (e.g., prothrombin fragments 1 + 2 and thrombin–antithrombin complex) and altered fibrinolytic activity
[14][61].
Interestingly, the activation of the coagulation system in AF may not be homogeneous throughout the body. Some studies have shown that platelet activation and thrombin generation markers were elevated in the atria of patients within minutes after AF induction, or as a consequence of rapid atrial pacing (RAP) in animal models, compared to peripheral circulation
[42][43][94,95]. These data highlight the effect of rhythm and rate on atrial pro-thrombotic mechanisms (e.g., local endocardial dysfunction/damage), which may promote a local pro-thrombotic environment.
Nevertheless, it is still not fully clarified whether “lone AF” (AF in the absence of apparent comorbidities) is sufficient to cause a pro-thrombotic state, or whether the presence of other underlying comorbidities and risk factors is required.
3. Activation of Coagulation Supports Atrial Cardiomyopathy
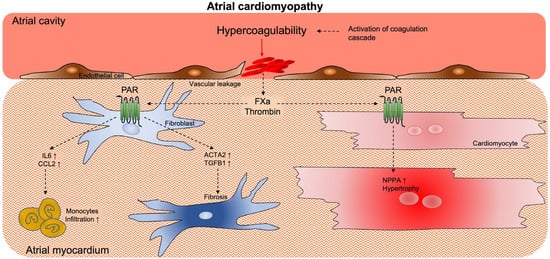
Activated coagulation factors, such as thrombin and FXa, can modulate physiological and pathological processes, such as inflammation and fibrosis, which may contribute to atrial cardiomyopathy (
Figure 3)
[44][45][102,103]. These extravascular (non-hemostatic) functions impact different cell types (e.g., endothelial cells, cardiomyocytes and cardiac fibroblasts) via the activation of protease-activated receptors (PAR)
[46][104]. The PAR family consists of four isoforms (PAR-1 to −4). Activated coagulation proteases, such as thrombin and FXa, cleave PAR at the N-terminus and generate an exposed N-tethered ligand that self-activates the receptor
[47][105].
Figure 3. Activation of coagulation promotes atrial cardiomyopathy. Activated coagulation factors, such as Thrombin and FXa, modulate cellular processes via the activation of PAR expressed on cardiac cells. These processes, such as inflammation, fibrosis and cellular hypertrophy, may contribute to the worsening of atrial cardiomyopathy. Abbreviations: PAR = protease-activated receptor; FXa = Factor × activated; IL6 = Interleukin 6; CCL2 = C-C motif ligand 2; NNPA = atrial natriuretic peptide.
Recently, hypercoagulability has been described to play a role in the progression of AF
[48][106]. The in vivo inhibition of FXa attenuated AF-induced atrial endomysial fibrosis and reduced AF complexity in goats after four weeks of AF
[48][106]. Several other studies have reported that the direct FXa inhibitor, rivaroxaban, attenuated cardiac fibrosis in various animal models of myocardial remodeling
[49][50][51][52][34,107,108,109].
To understand the mechanisms responsible for these effects,
scholarswe investigated the direct effect of activated coagulation factors, thrombin and FXa, on primary cardiac fibroblasts (CFs)
[53][110]. In this study, thrombin and FXa lead to the increased expression of well-known pro-fibrotic genes (e.g., Alpha 2 smooth muscle actin and Transforming growth factor beta genes) in CFs. Furthermore, FXa upregulated the gene expression of two key regulators of inflammatory processes, CCL2 and IL6, in primary adult human atrial CFs. This effect was mainly caused by FXa-induced PAR-1 activation, which was the most abundant isoform in CFs. Moreover, in line with previous findings,
thwe
scholars provided evidence for the existence of a positive feedback loop of PAR expression upon their activation by these coagulation factors
[48][53][106,110].
4. The Complex Association of AF and Thrombogenesis (Stroke)
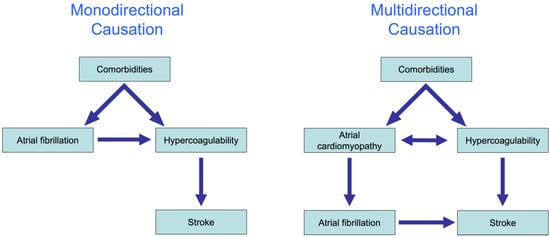
In recent years, the interaction between AF and stroke has been shown to be far more complicated than initially believed. The traditional hypothesis was that AF causes a reduction in the blood flow velocity, activation of coagulation factors in the blood and endothelial remodeling that in combination explain the enhanced risk for stroke in patients with AF. This hypothesis explains the association between AF and stroke largely by monodirectional causation from comorbidities to AF, to the activation of coagulation factors and ultimately to stroke (
Figure 4, left).
Figure 4. The complex association between AF and stroke. Monodirectional causation (
left): various comorbidities lead to the onset of AF, followed by the activation of the coagulation system, and ultimately stroke. Multidirectional causation (
right): atrial cardiomyopathy and hypercoagulability cause each other and share common pathophysiological pathways. These pathways, which may occur within and/or outside the atrial endothelium, can contribute to both proarrhythmic and prothrombotic mechanisms, resulting in the concomitant increased risk of AF and stroke.