Alzheimer’s disease (AD) is the most common neurodegenerative disorder in the elderly. The two cardinal neuropathological hallmarks of AD are the senile plaques, which are extracellular deposits mainly constituted by beta-amyloids, and neurofibrillary tangles formed by abnormally phosphorylated Tau (p-Tau) located in the cytoplasm of neurons.
- Alzheimer’s disease
- Tau protein
- neurofibrillary tangles
- amyloid-beta protein
1. Introduction
2. The Hallmark Lesions of AD: β-Amyloid and Tau Proteins
AD is characterized by neuron loss and increasing accumulation of neurofibrillary tangles formed by Tau protein inside the cells and the presence of amyloid plaques, mainly constituted by extracellularly aggregated amyloid beta-protein [9].2.1. Amyloid β-Peptide (Abeta)
Amyloid-β peptide (Abeta), ranging from 37 to 43 residues with different aggregation propensity, is obtained by the enzymatic cleavage of the Amyloid Precursor Protein (APP), a large transmembrane metal binding protein of 695–770 aminoacids [10,11][10][11]. The physiological role of APP is not yet fully understood. There are indications that the protein is involved in neurogenesis [12], neurite growth and long-term potentiation by regulation of calcium release [13]. It has been demonstrated that very small concentrations (picomolars) of Abeta improves memory in mice; while, on the contrary, high Abeta levels inhibit it [14]. Antimicrobial activity, inhibition of oncogenic viruses, enhanced activation of acetylcholine and nicotinic acetylcholine receptors have been observed as physiological effects of Abeta [15]. The cleavage of APP is the result of the activity of the enzymes of the secretase family, α- (ADAM), β- (BACE1) and γ- (or Presenilins) secretases, whose sequential intervention results in the onset of the amyloidogenic or non-amyloidogenic pathway (Figure 1).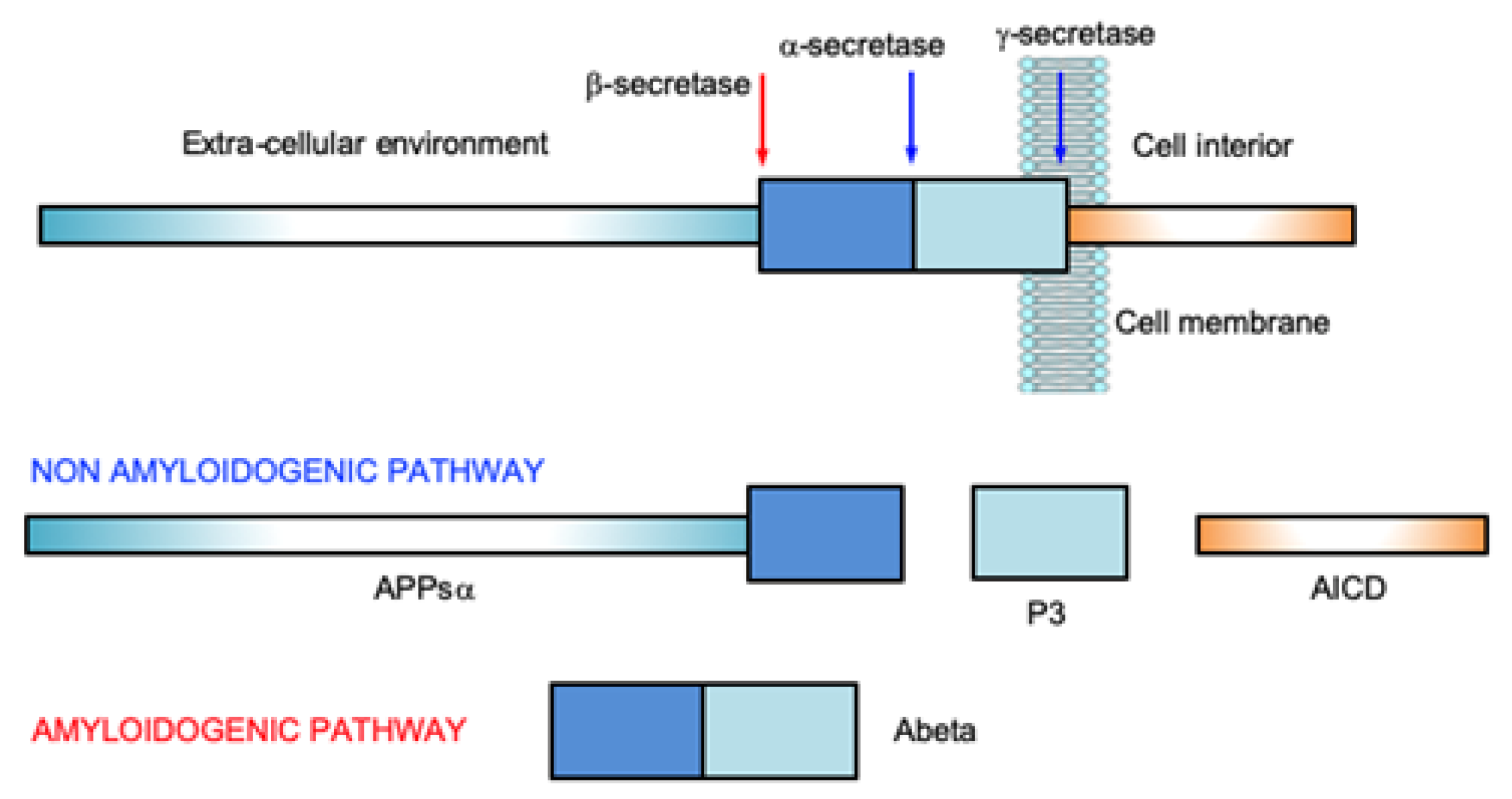
2.2. Tau Protein
Tau is a microtubule-associated protein, mainly expressed in the axons of neurons, deputed to mantain the microtubules that ensure the structural stability of the cell and allow the organelles, vesicles and proteins to move through the cell [38,39][29][30]. Several dysfunctions of Tau have been identified, constituting the family of neurogenerative diseases known as Tauopathies, including AD [40][31]. In solution, Tau possesses a harpin-like disordered and unfolded structure [39][30]. Because of different splicing during human MAPT gene translation, six different isoforms of Tau are present: 3R0N (352 aminoacids, aa.), 3R1N (381 aa.), 3R2N (410 aa.), 4R0N (383 aa.), 4R1N (412 aa.) and 4R2N (441 aa.), depending on the absence (0N) or presence of one (1N) or two (2N) inserts at the N terminus of the protein (Figure 3).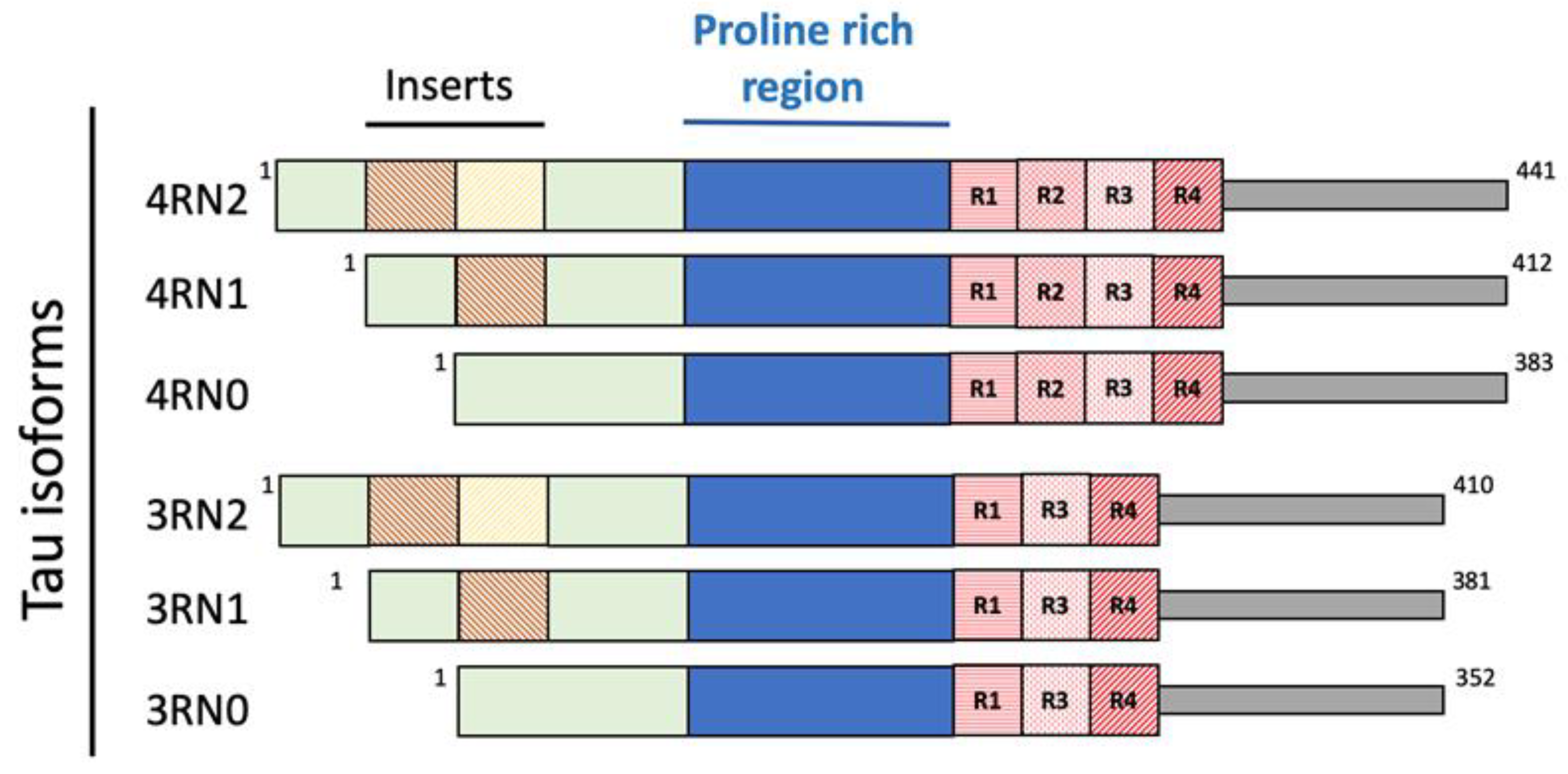
2.3. Proteins and Metal Ions in AD
2.4. Peptide-Based Scaffolds to Target Cu Ions as Therapeutics
Nanostructured peptides with metal binding properties are promising therapeutic advancements in neurodegenerative diseases. These nanostructures interact with metal ions and influence the biological properties of several proteins involved in neurodegenerative diseases [71][51]. The brain copper imbalance plays an important role in Abeta aggregation and in AD neurotoxicity. Moreover, the Cu2+ ion bound to Abeta can induce ROS production. Histidine-containing peptides and proteins are excellent metal binders and are found in many natural systems. For this aim, Caballero and Collaborators studied three short peptides, HWH, HKCH and HAH, forming highly stable albumin-like complexes, with higher affinity for Cu2+ than for Abeta(1–40). These copper-chelating peptides were designed with the aim of reducing copper toxicity in AD. Furthermore, HWH, HKCH and HAH act as very efficient inhibitors of copper-mediated generation of ROS and prevent the copper-induced overproduction of toxic oligomers in the early stages of amyloid aggregation in the presence of Cu2+ ions [60][43].3. Oxidative Stress and Its Involvement in AD Onset
Increasing evidence indicates that oxidative activity may be involved in the etiology of AD as well as other neurodegenerative pathologies and cancer. Under physiological conditions, free radicals, reactive oxygen species (ROS) and reactive nitrogen species (NOS), are normally produced in living cells; just consider, for example, the molecular species generated during the mitochondrial electron transport chain (ETC) and the Krebs cycle [75][52]. These unstable molecules, with unpaired electrons, initiate a series of reactions leading to the oxidation of proteins, lipids, and nucleic acids. However, in several cases and at low-to-moderate concentrations, free radicals play a physiological role [76][53]. ROS derived by the action of NADPH oxidase, a superoxide-oxidase enzyme, can fight the bacterial infection in the neutrophil phagosome [77][54]. Furthermore, ROS are physiologically involved in some cellular pathway signaling and in the regulation of the vascular tone, cell adhesion and apoptosis [76][53]. They also have a key role in the protection of adults and embryonic stem cells [78][55]. In healthy individuals, the excess production of free radical concentration is counteracted by the oxidative defense system, including glutathione, arginine, and citrulline; some chemical elements such as selenium and zinc; the vitamins A, C and E; the enzymes superoxide dismutase, catalase, glutathione reductase and glutathione peroxidases [79][56]. Aging and age-related diseases contribute to the free radical productions [80][57].4. The Antioxidant Properties of Egg-Derived Peptides
Proteins are huge biomolecular and macromolecular structures made up of one or more long chains of amino acid residues. Proteins serve a wide range of roles within animals, including catalyzing metabolic reactions, providing structure to cells and organisms, DNA replication, transporting chemicals, and responding to stimuli. A polypeptide is a linear chain of amino acid residues. Short polypeptides with fewer than 20–30 residues are rarely regarded as proteins and are often referred to as peptides; this is why peptides can be created by the enzymatic digestion of proteins.
Egg white-derived peptides, DHTKE (Asp-His-Thr-Lys-Glu), FFGFN (Phe-Phe-Glu-Phe-His), and MPDAHL (Met-Pro-Asp-Ala-His-Leu), formed via alcalase, were discovered to have antioxidant properties [92,94][58][59]. The egg white hydrolyzed by “protease P” give rise to two strongly antioxidant peptides, AEERYP (Ala-Glu-Glu-Arg-Tyr-Pro) and DEDTQAMP (Asp-Glu-Asp-Thr-Gln-Ala-Met-Pro). Pepsin hydrolyzed ovalbumin-derived peptide Tyr-Ala-Glu-Glu-Arg-Tyr-Pro-Ile-Leu has previously been reported to have angiotensin converting enzyme (ACE)-inhibitory activity and showed radical scavenging activity [92,95][58][60]. Two antioxidant tetrapeptides (Trp-Asn-Ile-Pro and Gly-Trp-Asn-Ile) were attained from the pyrolytic hydrolyzate of ovotransferrin [96][61]. Trp-Asn-Ile was proposed as a peptide motif involved in the significant activity of the above tetrapeptides. The ovotransferrin-derived tripeptide Ile-Arg-Trp exhibited powerful radical scavenging activity due to the tryptophan and the peptide bond between Trp and Arg [86,92][58][62]. Ovomucin-derived pentapeptide Trp-Asn-Trp-Ala-Asp has been found to decrease H2O2-induced oxidative stress in human fetal kidney cells (HEK-293) by hindering intracellular ROS accumulation. On the other side, from egg yolk, phosvitin phosphopeptides (PPP) obtained from tryptic digestion of phosvitin presented a protective effect against H2O2-induced oxidative stress in human intestinal epithelial cells [92][58] and, compared with intact phosvitin, PPP has shown a powerful ability to prevent lipid oxidation in the linoleic acid system and more efficient free radical capture [97][63].
5. Cholinesterase and BACE Inhibitory Activity of Egg-Derived Peptides
The cholinergic loss is one of the most prominent components of the neuropathology of Alzheimer’s disease. The cholinergic system is important for neuronal functions such as memory and learning by playing a main role in promoting neuronal plasticity. The cholinergic hypothesis considers that the level of acetylcholine in the brain of AD patients is low. This can happen because of the degradation produced by two cholinesterases: the first one is the true cholinesterase, AChE, and the other one is a pseudo-cholinesterase, BChE [98][64]. The hypothesis has received convincing validations, as AChE inhibitors are currently the most prescribed class of drugs for the treatment of AD [99][65].
Among the four peptides, KLPGF (at the concentration of 50 μg/mL) showed the greatest AChE and BChE inhibitory activity, with inhibition values of 61.23 ± 4.73% and 3.29 ± 0.93%, respectively. Peptide TNGIIR exhibited modest AChE and BChE inhibition with the value of 58.02 ± 1.89% and 1.50 ± 0.24%, respectively. Peptides QIGLF and RVPSL had no noteworthy AChE and BChE inhibitory properties. Furthermore, the peptide KLPGF made a number of powerful hydrogen bonds with numerous important amino acid residues situated in the catalytic and allosteric sites of AChE and a number of hydrophobic interactions with AChE. The contacts between KLPGF and AChE mostly involved the resulting amino acid residues: Tyr70-Trp84-Gly118-Gly119-Trp279-Asp285-Ser286-Ile287-Phe330-Phe331-Tyr334-His440-Gly441 [99][65].
6. Beta-Sheet Breaker (BSB) Peptides as Abeta Aggregation-Inhibitor
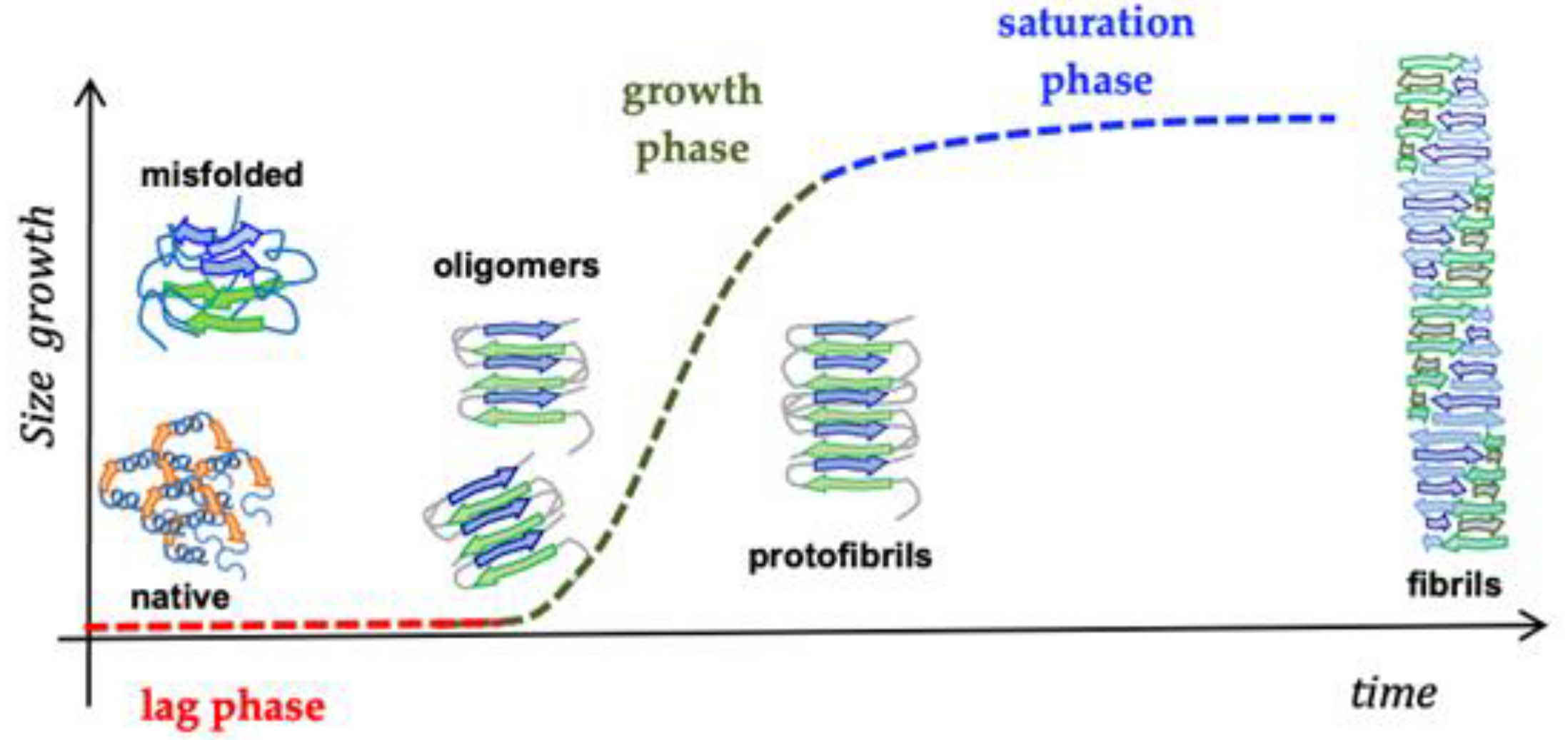
Significant evidence indicated that the key pathological event in Alzheimer’s disease is the switch from a normal soluble Abeta into beta-sheet-rich oligomeric structures which have the capacity to form insoluble amyloid deposits with neurotoxic effects in the brain. Thus, an attractive approach against AD is the inhibition of the aggregation of Abeta through the insertion of different-sized molecules able to prevent fibril formation [102][66]. Thus, several studies have been based on the design of a wide range of compounds, from small peptides to large chaperones, to develop inhibitors of Abeta aggregation [103,104][67][68]. In the late 1990s, Soto and coworkers reported the results of the in vitro addition of different concentrations of a five-residue synthetic peptide, called Beta-Sheet Breaker (BSB), in the solution containing Abeta40 molecules capable of impeding their aggregation [105][69]. BSBs represent a class of compounds intended to bind Abeta in specific ways to inhibit and/or block its pathological conformational modification and growth. There are several causes that trigger Abeta formation, and among these are pH changes, apolipoprotein E (ApoE), especially its E4 isoform [106][70], α1-antichymotrypsin [106][70], and C1q complement factor [107][71], oxidative stress [108][72], metals [109][73], and proteoglycans [110][74]. Many distinct small compounds have been shown to avoid and/or annul Abeta polymerization in vitro, unfortunately they lack specificity, a clear mechanism of action, and sometimes show high toxicity, making them difficult to improve and to clinically use [111][75]. Several studies have confirmed that different Abeta peptide regions contribute in a different way to aggregation and have shed light on several important interactions among specific peptide regions that control this process and are crucial for the peptide’s ability to aggregate and promote neurotoxicity. These regions are: the N-terminus (fragment 1–15) [112][76], the hydrophobic core (fragments 16–20) [113][77], the hinge or turn regions (fragments 22–27), [114][78] and the C-terminus (fragments 31–40/42) [115][79] (Figure 4).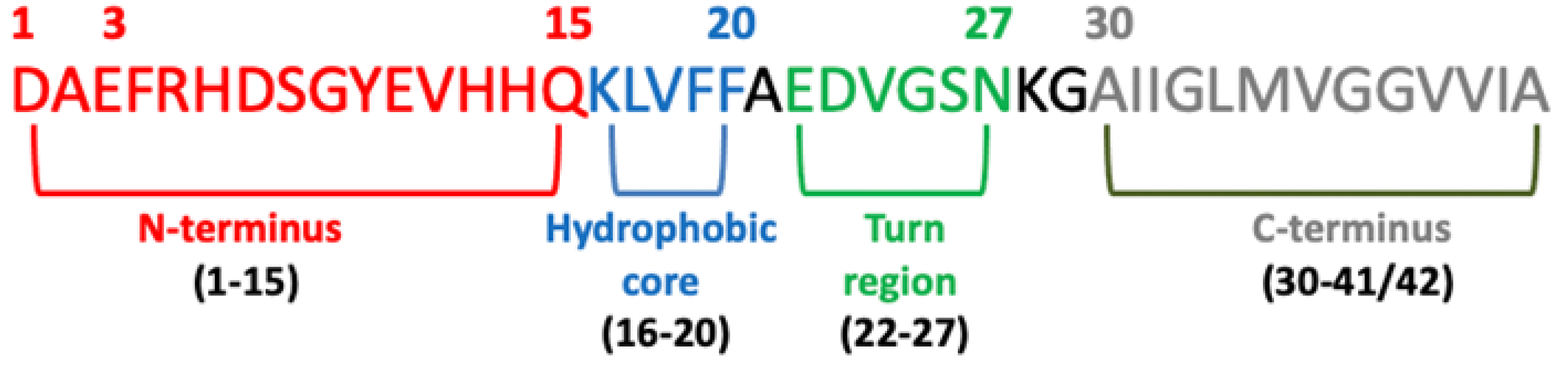
7. The Blood–Brain Barrier (BBB) and AD
The term blood–brain barrier describes the exclusive properties of the central nervous system microvasculature. These central nervous system vessels are non-fenestrated continuous vessels that contain some supplementary properties that allow them to tightly regulate the movement of cells, molecules and ions between the central nervous system and the blood [124]. Thus, BBB endothelial cells tightly regulate central nervous system homeostasis thanks to heavily restricting barrier capacity. This function is critical for proper neuronal function and to protect the central nervous system from injury, toxins, disease, pathogens, and inflammation [125]. The selective and restrictive proprieties of the BBB are an obstacle for drug delivery to the central nervous system. Today, the BBB is thought of as a complex and dynamic interface rather than as a static barrier [126].
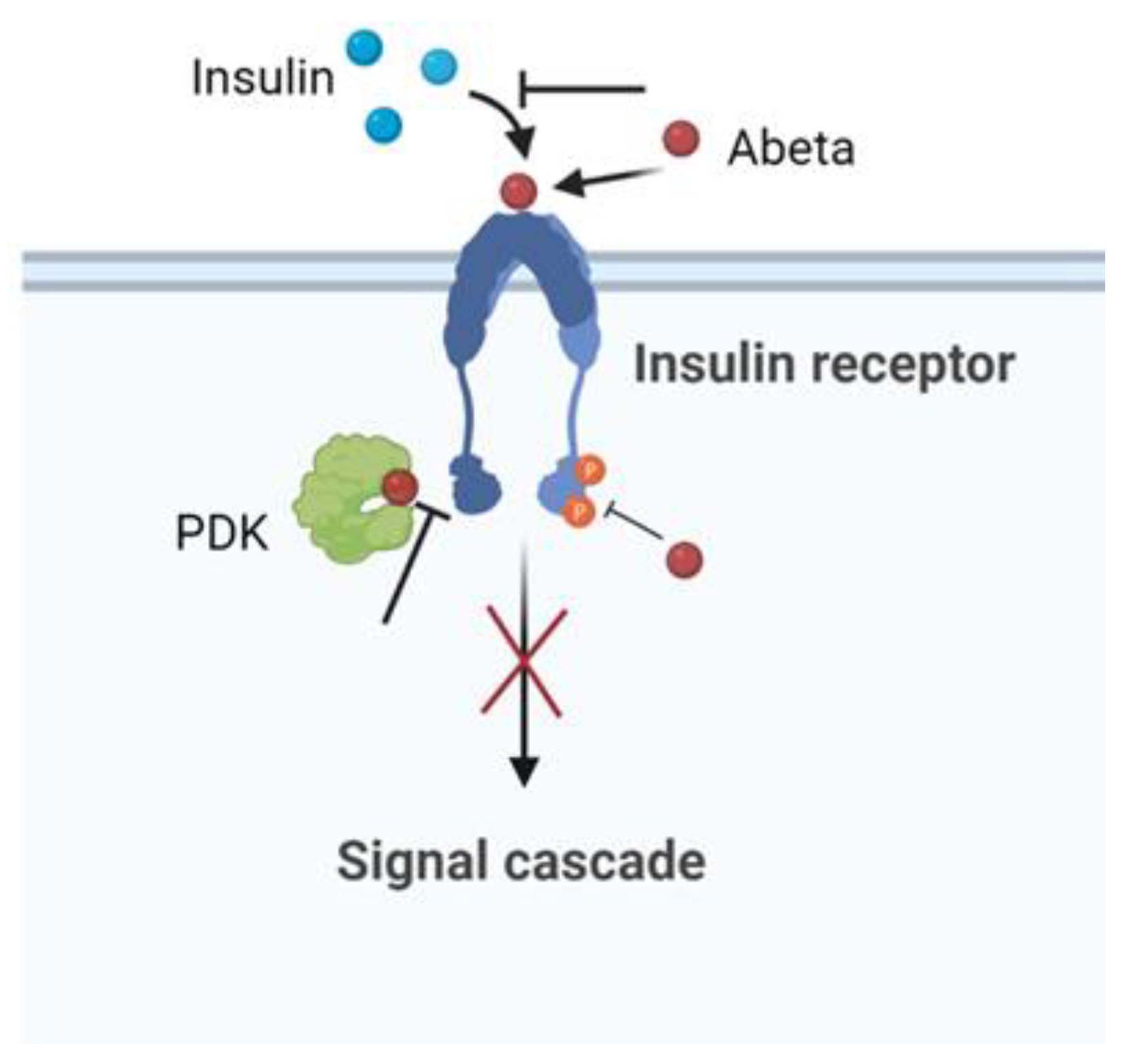
8. The Insulin Effect against AD
Insulin, peptide secreted by the pancreas, plays an important role in the regulation of the glucose metabolism in the peripheral tissues. The brain was once considered an insulin-insensitive organ, but today the insulin receptors are present throughout the brain and play a vital role for brain functioning [128]. Human and animal studies indicate that insulin influences cerebral bioenergetics, enhances synaptic viability and dendritic spine formation, increases the turnover of neurotransmitters, and modulates vascular function through effects on vasoreactivity, lipid metabolism, and inflammation [128].
7. The Blood–Brain Barrier (BBB) and AD
The term blood–brain barrier describes the exclusive properties of the central nervous system microvasculature. These central nervous system vessels are non-fenestrated continuous vessels that contain some supplementary properties that allow them to tightly regulate the movement of cells, molecules and ions between the central nervous system and the blood [80]. Thus, BBB endothelial cells tightly regulate central nervous system homeostasis thanks to heavily restricting barrier capacity. This function is critical for proper neuronal function and to protect the central nervous system from injury, toxins, disease, pathogens, and inflammation [81]. The selective and restrictive proprieties of the BBB are an obstacle for drug delivery to the central nervous system. Today, the BBB is thought of as a complex and dynamic interface rather than as a static barrier [82].
8. The Insulin Effect against AD
Insulin, peptide secreted by the pancreas, plays an important role in the regulation of the glucose metabolism in the peripheral tissues. The brain was once considered an insulin-insensitive organ, but today the insulin receptors are present throughout the brain and play a vital role for brain functioning [84]. Human and animal studies indicate that insulin influences cerebral bioenergetics, enhances synaptic viability and dendritic spine formation, increases the turnover of neurotransmitters, and modulates vascular function through effects on vasoreactivity, lipid metabolism, and inflammation [84].
9. Large-Size Proteins and AD
References
- Picone, P.; Di Carlo, M.; Nuzzo, D. Obesity and Alzheimer’s disease: Molecular bases. Eur. J. Neurosci. 2020, 52, 3944–3950.
- Raudino, F. Non-cognitive symptoms and related conditions in the Alzheimer’s Disease: A literature review. Neurol. Sci. 2013, 34, 1275–1282.
- Jessen, F.; Amariglio, R.E.; van Boxtel, M.; Breteler, M.; Ceccaldi, M.; Chételat, G.; Dubois, B.; Dufouil, C.; Ellis, K.A.; van der Flier, W.M.; et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement. 2014, 10, 844–852.
- Mariani, E.; Monastero, R.; Mecocci, P. Mild cognitive impairment: A systematic review. J. Alzheimers Dis. 2007, 12, 23–35.
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590.
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547.
- Delmotte, K.; Schaeverbeke, J.; Poesen, K.; Vandenberghe, R. Prognostic value of amyloid/tau/neurodegeneration (ATN) classification based on diagnostic cerebrospinal fluid samples for Alzheimer’s disease. Alzheimers Res. Ther. 2021, 13, 84.
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–284.
- Di Carlo, M.; Giacomazza, D.; San Biagio, P.L. Alzheimer’s Disease: Biological aspects, therapeutic perspectives and diagnostic tools. J. Phys. Condens. Matter 2012, 24, 244102.
- Chow, N.; Korenberg, J.R.; Chen, X.-N.; Neve, R.L. APP-BP1, a novel protein that binds to the carboxyl-terminal region of the Amyloid Precursor Protein. J. Biol. Chem. 1996, 271, 11339–11346.
- Wilquet, V.; De Strooper, B. Amyloid-beta precursor protein processing in neurodegeneration. Curr. Opin. Neurobiol. 2004, 14, 582–588.
- Dawkins, E.; Small, D.H. Insights into the physiological function of the β-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 2014, 129, 756–769.
- Morley, J.E.; Farr, S.A.; Nguyen, A.D.; Xu, F. What is the physiological function of amyloid-beta protein? J. Nutr. Health Aging 2019, 23, 225–226.
- Flood, J.F.; Morley, J.E.; Roberts, E. Amnestic effects in mice of four synthetic peptides homologous to amyloid beta protein from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 3363–3366.
- Pearson, H.A.; Peers, C. Physiological roles for amyloid β peptide. J. Physiol. 2006, 575, 5–10.
- Flammang, B.; Paradossi-Piquard, R.; Sevalle, J.; Bebayle, D.; Dabert-Gay, A.-S.; Thevenet, A.; Lauritzen, I.; Checler, F. Evidence that the amyloid-β protein precursor intracellular domain, AICD, derives from β-secretase-generated C-terminal fragment. J. Alzheimers Dis. 2012, 30, 145–153.
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270.
- Tanabe, C.; Hotoda, N.; Sasagawa, N.; Sehara-Fujisawa, A.; Maruyama, K.; Ishiura, S. ADAM19 is tightly associated with constitutive Alzheimer’s disease APP alpha-secretase in A172 cells. Biochem. Biophys. Res. Commun. 2006, 352, 111–117.
- Postina, R.; Schroeder, A.; Dewachter, I.; Bohl, J.; Schmitt, U.; Kojro, E.; Prinzen, C.; Endres, K.; Hiemke, C.; Blessing, M.; et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Investig. 2004, 113, 1456–1464.
- Haass, C. Take five–BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004, 23, 483–488.
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384.
- Coimbra, J.R.M.; Marques, D.F.F.; Baptista, S.J.; Pereira, C.M.F.; Moreira, P.I.; Dinis, T.C.P.; Santos, A.E.; Salvador, J.A.R. Highlights in BACE1 inhibitors for Alzheimer’s disease treatment. Front. Chem. 2018, 6, 178.
- Hansson, C.A.; Frykman, S.; Farmery, M.R.; Tjenberg, L.O.; Nilsberth, C.; Pursglove, S.E.; Ito, A.; Winblad, B.; Cowburn, R.F.; Thyberg, J. Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J. Biol. Chem. 2004, 279, 51654–51660.
- Buell, A.K. The growth of amyloid fibrils: Rates and mechanisms. Biochem. J. 2019, 476, 2677–2703.
- Deleanu, M.; Hernandez, J.-F.; Cippelletti, L.; Biron, J.P.; Rossi, E.; Taverna, M.; Cottet, H.; Chamieh, J. Unraveling the speciation of b-amyloid peptides during the aggregation process by Taylor dispersion analysis. Anal. Chem. 2021, 93, 6523–6533.
- Westermark, P.; Wernstedt, C.; Wilander, E.; Hayden, D.W.; O’Brian, T.D.; Johnson, K.H. Amyloid fibrils in human insulinoma and islets of Langherans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3881–3885.
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.-T.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet amyloid polypeptide: Structure, function, and pathology. J. Diab. Res. 2016, 2798269.
- Seeliger, J.; Weise, K.; Opitz, N.; Winter, R. The Effect of Aβ on IAPP Aggregation in the Presence of an Isolated β-Cell Membrane. J. Mol. Biol. 2012, 421, 348–363.
- Simic, G.; Babic Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milosevic, N.; Bazadona, D.; Buée, L.; da Silva, R.R.; Di Giovanni, G.; et al. Tau protein hyperphoshorilation and aggregation in Alzheimer’s Disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 6.
- Verwilst, P.; Kim, H.S.; Kim, S.; Kang, C.; Kim, J.S. Shedding light on tau protein aggregation: The progress in developing highly selective fluorophores. Chem. Soc. Rev. 2018, 47, 2249–2265.
- Hernandez, F.; Avila, J. Tauopathies. Cell. Mol. Life Sci. 2007, 64, 2219–2233.
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52.
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? ACC Chem. Res. 2019, 52, 2026–2035.
- Cristovao, J.S.; Santos, R.; Gomes, C.M. Metals and neuronal metal bindind proteins implicated in Alzheimer’s disease. Oxidative Med. Cell. Long. 2016, 2016, 9812178.
- Cecarini, V.; Gee, J.; Fioretti, E.; Amici, M.; Angeletti, M.; Eleuteri, A.M.; Keller, J.N. Protein oxidation and cellular homeostasis: Enphasis on metabolism. Biochim. Biophys. Acta 2007, 1773, 93–104.
- Shichiri, M. The role of lipid peroxidation in neurological disorders. J. Clin. Biochem. Nutr. 2014, 54, 151–160.
- Chen, W.-T.; Liao, Y.-H.; Yu, H.-M.; Cheng, I.H.; Chen, Y.-R. Distinct effects of Zn2+, Cu2+, Fe3+, and Al3+ on amyloid-β stability, oligomerization, and aggregation: Amyloid-β destabilization promotes annular protofibril formation. J. Biol. Chem. 2011, 286, 9646–9656.
- Baum, L.; Chan, I.H.S.; Cheung, S.K.-K.; Goggins, W.B.; Mok, V.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; Woo, J.; et al. Serum zinc is decreased in Alzheimer’s disease and serum arsenic correlates positively with cognitive ability. BioMetals 2010, 23, 173–179.
- Bishop, G.M.; Robinson, S.R.; Liu, Q.; Perry, G.; Atwood, C.S.; Smith, M.A. Iron: A pathological mediator of Alzheimer disease? Dev. Neurosci. 2002, 24, 184–187.
- Tougu, V.; Karafin, A.; Zovo, K.; Chung, R.S.; Howells, C.; West, A.K.; Palumaa, P. Zn(II)- and Cu(II)-induced non-fibrillar aggregates of amyloid-β (1–42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators. J. Neurochem. 2009, 110, 1784–1795.
- Sarell, C.J.; Wilkinson, S.R.; Viles, J.H. Substoichiometric levels of Cu2+ ions accelerate the kinetics of fiber formation and promote cell toxicity of amyloid-β from Alzheimer disease. J. Biol. Chem. 2010, 285, 41533–41540.
- Wild, K.; August, A.; Pietrzik, C.U.; Kins, S. Structure and synaptic function of metal binding to amyloid precursor protein and its proteolytic fragments. Front. Mol. Neurosci. 2017, 10, 21.
- Caballero, A.B.; Terol-Ordaz, L.; Espargaró, A.; Vázquez, G.; Nicolás, E.; Sabaté, R.; Gamez, P. Histidine-Rich Oligopeptides to Lessen Copper-Mediated Amyloid-β Toxicity. Chemistry 2016, 22, 7268–7280.
- Esmieu, C.; Ferrand, G.; Borghesani, V.; Hureau, C. Impact of N-Truncated Aβ Peptides on Cu- and Cu(Aβ)-Generated ROS: Cu I Matters! Chemistry 2021, 27, 1777–1786.
- Koh, J.-Y.; Lee, S.-J. Metallothionein-3 as a multifunctional player in the control of cellular processes and diseases. Mol. Brain 2020, 13, 116.
- Atrián-Blasco, E.; Santoro, A.; Pountney, D.L.; Meloni, G.; Hureau, C.; Faller, P. Chemistry of mammalian metallothioneins and their interaction with amyloidogenic peptides and proteins. Chem. Soc. Rev. 2017, 46, 7683–7693.
- Xu, W.; Xu, Q.; Cheng, H.; Tan, X. The efficacy and pharmacological mechanism of Zn7MT3 to protect against Alzheimer’s disease. Sci. Rep. 2017, 7, 13763.
- Cristovao, J.S.; Gomes, C.M. S100 proteins in Alzheimer’s disease. Front. Neurosci. 2019, 13, 463.
- McAllister, B.B.; Dyck, R.H. Zinc transporter 3 (ZnT3) and vescicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017, 80, 329–350.
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1636.
- Trapani, G.; Satriano, C.; La Mendola, D. Peptides and their Metal Complexes in Neurodegenerative Diseases: From Structural Studies to Nanomedicine Prospects. Curr. Med. Chem. 2018, 25, 715–747.
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial respiratory complex I: Structure, function and implication in human diseases. Curr. Med. Chem. 2009, 16, 1266–1277.
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human diseases. Int. J. Biochem. Cell Biol. 2007, 39, 44–84.
- Rada, B.; Leto, T.L. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib. Microbiol. 2008, 15, 164–187.
- Sinenko, S.A.; Starkova, T.Y.; Kurzmin, A.A.; Tomilin, A.N. Physiological signaling functions of reactive oxygen species in stem cells: From flies to man. Front. Cell Dev. Biol. 2021, 9, 714370.
- Di Carlo, M.; Giacomazza, D.; Picone, P.; Nuzzo, D.; San Biagio, P.L. Are oxidative stress and mitochondrial dysfunction the key players in the neurodegenerative diseases? Free Ras. Res. 2012, 46, 1327–1338.
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging and diseases. Clin. Interv. Aging 2018, 13, 757–772.
- Nimalaratne, C.; Wu, J. Hen egg as an antioxidant food commodity: A review. Nutrients 2015, 7, 8274–8293.
- Liu, J.; Jin, Y.; Lin, S.; Jones, G.S.; Chen, F. Purification and identification of novel antioxidant peptides from egg white protein and their antioxidant activities. Food Chem. 2015, 175, 258–266.
- Davalos, A.; Miguel, M.; Bartolomé, B.; López-Fandiño, R. Antioxidant activity of peptides derived from egg white proteins by enzymatic hydrolysis. J. Food Prot. 2004, 67, 1939–1944.
- Huang, W.; Shen, S.; Nimalaratne, C.; Li, C.; Majumder, K.; Wu, J. Effects of addition of egg ovotransferrin-derived peptides on the oxygen radical absorbance capacity of different teas. Food Chem. 2012, 135, 1600–1607.
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464.
- Xu, X.; Katayama, S.; Mine, Y. Antioxidant activity of tryptic digests of hen egg yolk phosvitin: Antioxidant activity of phosvitin peptides. J. Sci. Food Agric. 2007, 87, 2604–2608.
- Yu, Z.; Dong, W.; Wu, S.; Shen, J.; Zhao, W.; Ding, L.; Liu, J.; Zheng, F. Identification of ovalbumin-derived peptides as multi-target inhibitors of AChE, BChE, and BACE1. J. Sci. Food Agric. 2020, 100, 2648–2655.
- Yu, Z.; Wu, S.; Zhao, W.; Ding, L.; Fan, Y.; Shiuan, D.; Liu, J.; Chen, F. Anti-Alzheimers activity and molecular mechanism of albumin-derived peptides against AChE and BChE. Food Funct. 2018, 9, 1173–1178.
- Mason, J.M.; Kokkoni, N.; Stott, K.; Doig, A.J. Design strategies for anti-amyloid agents. Curr. Opin. Struct. Biol. 2003, 13, 526–532.
- Goyal, D.; Shuaib, S.; Mann, S.; Goyal, B. Rationally designed peptides and peptidomimetics as inhibitors of amyloid-β (Ab) aggregation: Potential therapeutics of Alzheimer’s Disease. ACS Comb. Sci. 2017, 19, 55–80.
- Belluti, F.; Rampa, A.; Gobbi, S.; Bisi, A. Small-molecule inhibitors/modulators of amyloid-β peptide aggregation and toxicity for the treatment of Alzheimer’s disease: A patent review (2010–2012). Expert Opin. Ther. Pat. 2013, 23, 581–596.
- Soto, C.; Kindy, M.S.; Baumann, M.; Frangione, B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent β-sheet conformation. Biochem. Biophys. Res. Comm. 1996, 116, 672–680.
- Ma, J.; Yee, A.; Brewer, H.B., Jr.; Das, S.; Potter, H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 1994, 372, 92–94.
- Boyett, K.W.; DiCarlo, G.; Jantzen, P.T.; Jackson, J.; O’Leary, C.; Wilcock, D.; Morgan, D.; Gordon, M.N. Increased fibrillar beta-amyloid in response to human C1q injections into hippocampus and cortex of APP + PS1 transgenic mice. Neurochem. Res. 2003, 28, 83–93.
- Komatsu, H.; Liu, L.; Murray, I.V.J.; Axelsen, P.H. A mechanistic link between oxidative stress and membrane mediated amyloidogenesis revealed by infrared spectroscopy. Biochim. Biophys. Acta-Biomembr. 2007, 1768, 1913–1922.
- Stellato, F.; Fusco, Z.; Chiaraluce, R.; Consalvi, V.; Dinarelli, S.; Placidi, E.; Petrosino, M.; Rossi, G.C.; Minicozzi, V.; Morante, S. The effect of β-sheet breaker peptides on metal associated Amyloid-β peptide aggregation process. Biophys. Chem. 2017, 229, 110–114.
- Snow, A.D.; Wight, T.N. Proteoglycans in the pathogenesis of Alzheimer’s disease and other amyloidosis. Neurobiol. Aging 1989, 10, 481–497.
- Walsh, D.M.; Selkoe, D.J. Abeta oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184.
- Gardberg, A.S.; Dice, L.T.; Ou, S.; Rich, R.L.; Helmbrecht, E.; Ko, J.; Wetzel, R.; Myszka, D.G.; Patterson, P.H.; Dealwis, C. Molecular basis for passive immunotherapy of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 15659–15664.
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid fibrils of the HET-s (218–289) prion form a beta solenoid with a triangular hydrophobic core. Science 2008, 319, 1523–1526.
- Maji, S.K.; Ogorzalek Loo, R.R.; Inayathullah, M.; Spring, S.M.; Vollers, S.S.; Condron, M.M.; Bitan, G.; Loo, J.A.; Teplow, D.B. Amino acid position-specific contributions to amyloid beta-protein oligomerization. J. Biol. Chem. 2009, 284, 23580–23591.
- Fradinger, E.A.; Monien, B.H.; Urbanc, B.; Lomakin, A.; Tan, M.; Li, H.; Spring, S.M.; Condron, M.M.; Cruz, L.; Xie, L.C.; et al. C-terminal peptides coassemble into Abeta42 oligomers and protect neurons against Abeta42-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 14175–14180.
- Villabona-Rueda, A.; Erice, C.; Pardo, C.A.; Stins, M.F. The evolving concept pf the blood brain barrier (BBB): From a single static barrier to a heterogeneous and dynamic relay center. Front. Cell. Neurosci. 2019, 13, 405.
- Daneman, R.; Prat, A. The Blood-Brain Barrier. Cold Spring Harbor. Perspect. Biol. 2015, 7, a020412.
- Schofield, C.L.; Rodrigo-Navarro, A.; Dalby, M.J.; Van Agtmael, T.; Salmeron-Sanchez, M. Biochemical- and biophysical-induced barrigenesis in the blood-brain barrier: A review of barrigenic factor for use in vitro models. Adv. NanoBiomed. Res. 2021, 1, 2000068.
- Wang, D.; Chen, F.; Han, Z.; Yin, Z.; Ge, X.; Lei, P. Relationship between amyloid-β deposition and blood-brain barrier dysfunction in Alheimer’s disease. Front. Cell. Neurosci. 2021, 15, 695479.
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243.
- Nuzzo, D.; Picone, P.; Baldassano, S.; Caruana, L.; Messina, E.; Marino Gammazza, A.; Cappello, F.; Mulè, F.; Di Carlo, M. Insulin resistance as common molecular denominator linking obesity to Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 723–735.
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766.
- Accardi, G.; Caruso, C.; Colonna-Romano, G.; Camarda, C.; Monastero, R.; Candore, G. Can Alzheimer disease be a form of type 3 diabetes? Rejuvenation Res. 2012, 15, 217–221.
- Willette, A.A.; Johnson, S.C.; Birdsill, A.C.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P.; et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement. 2015, 11, 504–510.e1.
- Ekblad, L.L.; Johansson, J.; Helin, S.; Viitanen, M.; Laine, H.; Puukka, P.; Jula, A.; Rinne, J.O. Midlife insulin resistance, APOE genotype, and late-life brain amyloid accumulation. Neurology 2018, 90, e1150–e1157.
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80.
- Zhao, W.-Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260.
- Lee, H.-K.; Kumar, P.; Fu, Q.; Rosen, K.M.; Querfurth, H.W. The insulin/AKT signaling pathway is targeted by intracellular β-amyloid. Mol. Biol. Cell 2009, 20, 1533–1544.
- Zhao, W.-Q.; Townsend, M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta-Mol. Basis Disease 2009, 1792, 482–496.
- Hendrick, J.P.; Hartl, F.U. Molecular chaperone functions of Heat-Shock Proteins. Ann. Rev. Biochem. 1993, 62, 349–384.
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In Vivo aspects of protein folding and quality control. Science 2016, 353, aac4354.
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. Recent advances in understanding catalysis of protein folding by molecular chaperones. FEBS Lett. 2020, 594, 2770–2781.
- Fink, A.L. Chaperone-mediated protein folding. Physiol. Rev. 1993, 79, 425–449.
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642.
- Fan, C.Y.; Lee, S.; Cyr, D.M. Mechanisms for regulating Hsp70 function by Hsp40. Cell Stress Chaperon 2003, 8, 309–316.
- Frank, E.; Madsen, O.; van Rheede, T.; Ricard, J.; Huynen, M.A.; de Jong, W.W. Evolutionary diversity of vertebrate small heat shock proteins. J. Mol. Evol. 2004, 59, 792–805.
- Stengel, F.; Baldwin, A.J.; Painter, A.J.; Benesh, J.L.P. Quaternary dynamics and plasticity underlie small heat shock protein chaperone function. Proc. Natl. Acad. Sci. USA 2010, 107, 2007–2012.
- Young, J.C.; Moarefi, I.; Hartl, F.U. Hsp90: A specialized but essential protein-folding tool. J. Cell Biol. 2001, 154, 267–273.
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 chaperones machines. Cell 1998, 92, 351–366.
- Mogk, A.; Bukau, B.; Kampinga, H.H. Cellular handling of protein aggregates by disaggregation machines. Mol. Cell. 2018, 69, 214–226.
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11–22.
- Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. USA 2012, 109, 12479–12484.
- Chaari, A. Molecular chaperones biochemistry and role in neurodegenerative diseases. Int. J. Biol. Macromol. 2019, 131, 396–411.
- Tittelmeier, J.; Nachman, E.; Nussbaum-Krammer, C. Molecular chaperones: A double-edged sword in neurodegenerative diseases. Front. Aging Neurosci. 2020, 12, 581374.
- Bahr, T.; Katuri, J.; Liang, T.; Bai, Y. Mitochondrial chaperones in human health and disease. Free Rad. Biomed. 2021, 179, 363–374.
- Nachman, E.; Wentink, A.S.; Madiona, K.; Bousset, L.; Katsinelos, T.; Allison, K.; Kampinga, H.; McEwan, W.A.; Jahn, T.R.; Melki, R.; et al. Disassembly of Tau fibrils by the human Hsp70 disaggregation machinery generates small seeding competent species. J. Biol. Chem. 2020, 295, 9676–9690.
- Ring, J.; Tadic, J.; Ristic, S.; Poglisch, M.; Bergmann, M.; Radic, N.; Mossmann, D.; Liang, Y.T.; Maglione, M.; Jerkovic, A.; et al. The Hsp40 chaperone Ydj1 drives amiloyd beta 42 toxicity. EMBO Mol. Med. 2022, 14, e13952.
- Arosio, P.; Michaels, T.C.T.; Linse, S.; Mansson, C.; Emanuelsson, C.; Presto, J.; Johansson, J.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 2016, 7, 10948.