Hepacivirus C (HCV) is a small (55–65 nm in size), enveloped, positive-sense single-stranded RNA virus of the family Flaviviridae. The Hepacivirus C is the cause of hepatitis C and some cancers such as liver cancer (hepatocellular carcinoma, abbreviated HCC) and lymphomas in humans.
- flaviviridae
- liver cancer
- hepacivirus
1. Taxonomy
The hepatitis C virus belongs to the genus Hepacivirus, a member of the family Flaviviridae. Until recently it was considered to be the only member of this genus. However a member of this genus has been discovered in dogs: canine hepacivirus.[1] There is also at least one virus in this genus that infects horses.[2] Several additional viruses in the genus have been described in bats and rodents.[3][4]
2. Structure

The hepatitis C virus particle consists of a lipid membrane envelope that is 55 to 65 nm in diameter.[5][6] Two viral envelope glycoproteins, E1 and E2, are embedded in the lipid envelope.[7] They take part in viral attachment and entry into the cell.[5] Within the envelope is an icosahedral core that is 33 to 40 nm in diameter.[6] Inside the core is the RNA material of the virus.[5]
E1 and E2 Glycoproteins
E1 and E2 are covalently bonded when embedded in the envelope of HCV and are stabilized by disulfide bonds. E2 is globular and seems to protrude 6 nm out from the envelope membrane according to electron microscope images.[6]
These glycoproteins play an important role in the interactions hepatitis C has with the immune system. The hypervariable region 1 (HVR1) can be found on the E2 glycoprotein.[5] HVR1 is flexible and quite accessible to surrounding molecules.[8] HVR1 helps E2 shield the virus from the immune system. It prevents CD81 from latching onto its respective receptor on the virus.[8] In addition, E2 can shield E1 from the immune system.[8] Although HVR1 is quite variable in amino acid sequence, this region has similar chemical, physical, and conformational characteristics across many E2 glycoproteins.[9]
3. Genome

Hepatitis C virus has a positive sense single-stranded RNA genome. The genome consists of a single open reading frame that is 9600 nucleotide bases long.[10] This single open reading frame is translated to produce a single protein product, which is then further processed to produce smaller active proteins. This is why on publicly available databases, such as the European Bioinformatic Institute, the viral proteome only consists of 2 proteins.
At the 5' and 3' ends of the RNA are the UTR, that are not translated into proteins but are important to translation and replication of the viral RNA. The 5' UTR has a ribosome binding site[11] or internal ribosome entry site (IRES) that initiates the translation of a very long protein containing about 3,000 amino acids. The core domain of the HCV IRES contains a four-way helical junction that is integrated within a predicted pseudoknot.[12] The conformation of this core domain constrains the open reading frame's orientation for positioning on the 40S ribosomal subunit. The large pre-protein is later cleaved by cellular and viral proteases into the 10 smaller proteins that allow viral replication within the host cell, or assemble into the mature viral particles.[13] Structural proteins made by the hepatitis C virus include Core protein, E1 and E2; nonstructural proteins include NS2, NS3, NS4A, NS4B, NS5A, and NS5B.
4. Molecular Biology

The proteins of this virus are arranged along the genome in the following order: N terminal-core-envelope (E1)–E2–p7-nonstructural protein 2 (NS2)–NS3–NS4A–NS4B–NS5A–NS5B–C terminal. The mature nonstructural proteins (NS2 to NS5B) generation relies on the activity of viral proteinases.[14] The NS2/NS3 junction is cleaved by a metal dependent autocatalytic proteinase encoded within NS2 and the N-terminus of NS3. The remaining cleavages downstream from this site are catalysed by a serine proteinase also contained within the N-terminal region of NS3.
The core protein has 191 amino acids and can be divided into three domains on the basis of hydrophobicity: domain 1 (residues 1–117) contains mainly basic residues with two short hydrophobic regions; domain 2 (residues 118–174) is less basic and more hydrophobic and its C-terminus is at the end of p21; domain 3 (residues 175–191) is highly hydrophobic and acts as a signal sequence for E1 envelope protein.
Both envelope proteins (E1 and E2) are highly glycosylated and important in cell entry. E1 serves as the fusogenic subunit and E2 acts as the receptor binding protein. E1 has 4–5 N-linked glycans and E2 has 11 N-glycosylation sites.
The p7 protein is dispensable for viral genome replication but plays a critical role in virus morphogenesis. This protein is a 63 amino acid membrane spanning protein which locates itself in the endoplasmic reticulum. Cleavage of p7 is mediated by the endoplasmic reticulum's signal peptidases. Two transmembrane domains of p7 are connected by a cytoplasmic loop and are oriented towards the endoplasmic reticulum's lumen.
NS2 protein is a 21–23 kiloDalton (kDa) transmembrane protein with protease activity.
NS3 is 67 kDa protein whose N-terminal has serine protease activity and whose C-terminal has NTPase/helicase activity. It is located within the endoplasmic reticulum and forms a heterodimeric complex with NS4A—a 54 amino acid membrane protein that acts as a cofactor of the proteinase.
NS4B is a small (27 kDa) hydrophobic integral membrane protein with 4 transmembrane domains. It is located within the endoplasmic reticulum and plays an important role for recruitment of other viral proteins. It induces morphological changes to the endoplasmic reticulum forming a structure termed the membranous web.
NS5A is a hydrophilic phosphoprotein which plays an important role in viral replication, modulation of cell signaling pathways and the interferon response. It is known to bind to endoplasmic reticulum anchored human VAP proteins.[15]
The NS5B protein (65 kDa) is the viral RNA dependent RNA polymerase. NS5B has the key function of replicating the HCV’s viral RNA by using the viral positive RNA strand as its template and catalyzes the polymerization of ribonucleoside triphosphates (rNTP) during RNA replication.[16][17][18] Several crystal structures of NS5B polymerase in several crystalline forms have been determined based on the same consensus sequence BK (HCV-BK, genotype 1).[19] The structure can be represented by a right hand shape with fingers, palm, and thumb. The encircled active site, unique to NS5B, is contained within the palm structure of the protein. Recent studies on NS5B protein genotype 1b strain J4’s (HC-J4) structure indicate a presence of an active site where possible control of nucleotide binding occurs and initiation of de-novo RNA synthesis. De-novo adds necessary primers for initiation of RNA replication.[20] Current research attempts to bind structures to this active site to alter its functionality in order to prevent further viral RNA replication.[21]
An 11th has also been described.[22][23] This protein is encoded by a +1 frameshift in the capsid gene. It appears to be antigenic but its function is unknown.
5. Replication
Replication of HCV involves several steps. The virus replicates mainly in the hepatocytes of the liver, where it is estimated that daily each infected cell produces approximately fifty virions (virus particles) with a calculated total of one trillion virions generated. The virus may also replicate in peripheral blood mononuclear cells, potentially accounting for the high levels of immunological disorders found in chronically infected HCV patients. In the liver, the HCV particles are brought into the sinusoids by blood flow. These sinusoids neighbor hepatocyte cells.[5] HCV is able to pass through the endothelium of the sinusoids and make its way to the basolateral surface of the hepatocyte cells.[5]
HCV has a wide variety of genotypes and mutates rapidly due to a high error rate on the part of the virus' RNA-dependent RNA polymerase. The mutation rate produces so many variants of the virus it is considered a quasispecies rather than a conventional virus species.[24] Entry into host cells occur through complex interactions between virions, especially through their glycoproteins, and cell-surface molecules CD81, LDL receptor, SR-BI, DC-SIGN, Claudin-1, and Occludin.[25][26]
The envelope of HCV is similar to very-low density lipoproteins (VLDL) and low-density lipoproteins (LDL).[5] Because of this similarity, the virus is thought to be able to associate with apolipoproteins. It could surround itself with lipoproteins, partially covering up E1 and E2. Recent research indicates that these apolipoproteins interact with scavenger receptor B1 (SR-B1). SR-B1 is able to remove lipids from the lipoproteins around the virus to better allow for HVR1 contact. Claudin 1, which is a tight-junction protein, and CD81 link to create a complex, priming them for later HCV infection processes. As immune system is triggered, macrophages increase the amount of TNF- α around the hepatocytes which are being infected. This triggers the migration of occludin, which is another tight-junction complex, to the basolateral membrane. The HCV particle is ready to enter the cell.[5]
These interactions lead to the endocytosis of the viral particle. This process is aided by clathrin proteins. Once inside an early endosome, the endosome and the viral envelope fuse and the RNA is allowed into the cytoplasm.[5]
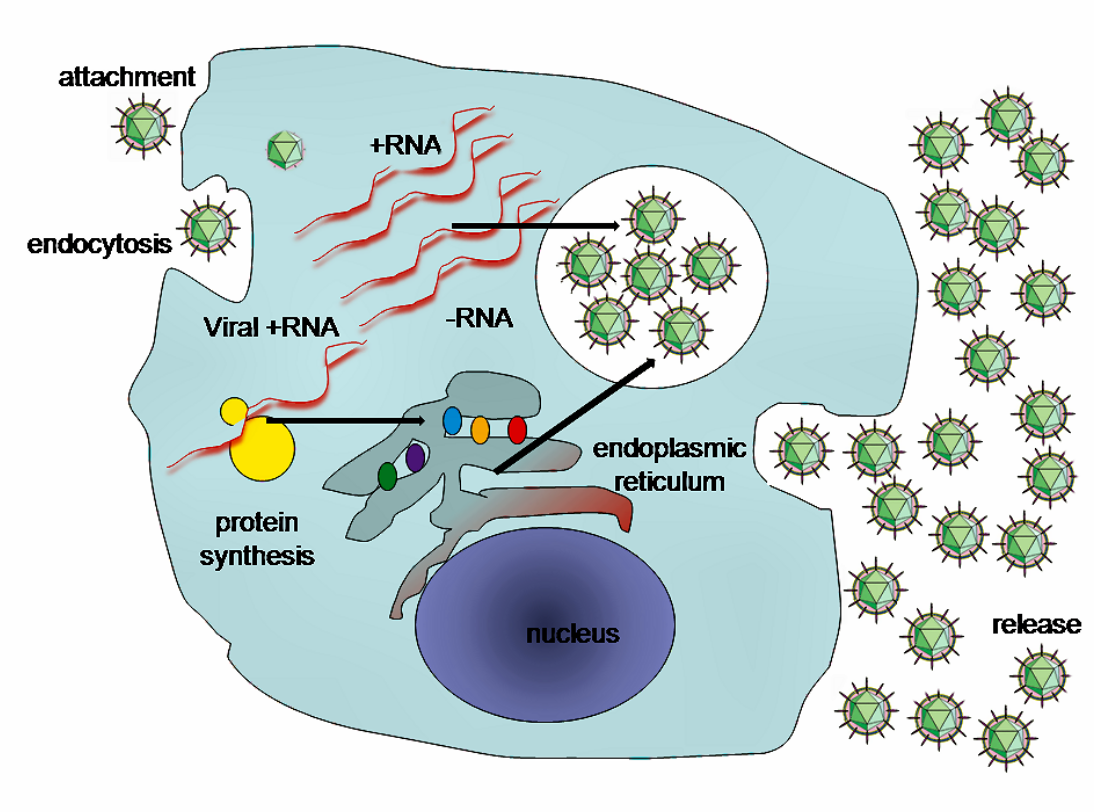
HCV takes over portions of the intracellular machinery to replicate.[27] The HCV genome is translated to produce a single protein of around 3011 amino acids. The polyprotein is then proteolytically processed by viral and cellular proteases to produce three structural (virion-associated) and seven nonstructural (NS) proteins. Alternatively, a frameshift may occur in the Core region to produce an Alternate Reading Frame Protein (ARFP).[28] HCV encodes two proteases, the NS2 cysteine autoprotease and the NS3-4A serine protease. The NS proteins then recruit the viral genome into an RNA replication complex, which is associated with rearranged cytoplasmic membranes. RNA replication takes places via the viral RNA-dependent RNA polymerase NS5B, which produces a negative strand RNA intermediate. The negative strand RNA then serves as a template for the production of new positive strand viral genomes. Nascent genomes can then be translated, further replicated or packaged within new virus particles.
The virus replicates on intracellular lipid membranes.[29] The endoplasmic reticulum in particular are deformed into uniquely shaped membrane structures termed 'membranous webs'. These structures can be induced by sole expression of the viral protein NS4B.[30] The core protein associates with lipid droplets and utilises microtubules and dyneins to alter their location to a perinuclear distribution.[31] Release from the hepatocyte may involve the very low density lipoprotein secretory pathway.[32] Another hypothesis states that the viral particle may be secreted from the endoplasmic reticulum through the endosomal-sorting complex required for transport (ESCRT) pathway.[5] This pathway is normally utilized to bud vesicles out of the cell. The only limitation to this hypothesis is that the pathway is normally used for cellular budding, and it is not known how HCV would commandeer the ESCRT pathway for use with the endoplasmic reticulum.[5]
6. Genotypes
Based on genetic differences between HCV isolates, the hepatitis C virus species is classified into six genotypes (1–6) with several subtypes within each genotype (represented by lower-cased letters).[33][34] Subtypes are further broken down into quasispecies based on their genetic diversity. Genotypes differ by 30–35% of the nucleotide sites over the complete genome.[35] The difference in genomic composition of subtypes of a genotype is usually 20–25%. Subtypes 1a and 1b are found worldwide and cause 60% of all cases.
Clinical importance
Genotype is clinically important in determining potential response to interferon-based therapy and the required duration of such therapy. Genotypes 1 and 4 are less responsive to interferon-based treatment than are the other genotypes (2, 3, 5 and 6).[36] The duration of standard interferon-based therapy for genotypes 1 and 4 is 48 weeks, whereas treatment for genotypes 2 and 3 is completed in 24 weeks. Sustained virological responses occur in 70% of genotype 1 cases, ~90% of genotypes 2 and 3, ~65% of genotype 4 and ~80% of genotype 6.[37] In addition people of African descent are much less likely to clear the infection when infected with genotypes 1 or 4 [38] and substantial proportion of this lack of response to treatment has been traced down to a single nucleotide polymorphism (SNP) on chromosome 19 that is predictive of treatment success.[39] HCV genotypes 1 and 4 have been distributed endemically in overlapping areas of West and Central Africa, infecting for centuries human populations carrying the genetic polymorphism in question. This has prompted scientists to suggest that the protracted persistence of HCV genotypes 1 and 4 in people of African origin is an evolutionary adaptation over many centuries to these populations’ immunogenetic responses.[40]
Infection with one genotype does not confer immunity against others, and concurrent infection with two strains is possible. In most of these cases, one of the strains removes the other from the host in a short time. This finding opens the door to replacing strains non-responsive to medication with others easier to treat.[41]
7. Epidemiology
Hepatitis C virus is predominantly a blood-borne virus, with very low risk of sexual or vertical transmission.[42] Because of this mode of spread the key groups at risk are intravenous drug users (IDUs), recipients of blood products and sometimes patients on haemodialysis. Common setting for transmission of HCV is also intra-hospital (nosocomial) transmission, when practices of hygiene and sterilization are not correctly followed in the clinic.[43] A number of cultural or ritual practices have been proposed as a potential historical mode of spread for hepatitis C virus, including circumcision, genital mutilation, ritual scarification, traditional tattooing and acupuncture.[42] It has also been argued that given the extremely prolonged periods of persistence of HCV in humans, even very low and undetectable rates of mechanical transmission via biting insects may be sufficient to maintain endemic infection in the tropic, where people receive large number of insect bites.[44]
8. Evolution
Identification of the origin of this virus has been difficult but genotypes 1 and 4 appear to share a common origin.[45] A Bayesian analysis suggests that the major genotypes diverged about 300–400 years ago from the ancestor virus.[46] The minor genotypes diverged about 200 years ago from their major genotypes. All of the extant genotypes appear to have evolved from genotype 1 subtype 1b.
A study of genotype 6 strains suggests an earlier date of evolution: ∼1,100 to 1,350 years before the present (95% credible region, 600 to >2,500 years ago).[47] The estimated rate of mutation was 1.8 × 10−4 (95% credible region 0.9 × 10−4 to 2.9 × 10−4). An experimental study estimated the mutation rate at 2.5 - 2.9 × 10-3 base substitutions/site/year.[48] This genotype may be the ancestor of the other genotypes.[47]
A study of European, USA and Japanese isolates suggested that the date of origin of genotype 1b was ~1925.[49] The estimated dates of origin of types 2a and 3a were 1917 and 1943 respectively. The time of divergence of types 1a and 1b was estimated to be 200–300 years.
A study of genotype 1a and 1b estimated the dates of origin to be 1914–1930 (95% credible interval: 1802–1957) for type 1a and 1911–1944 (95% credible interval: 1806–1965) for type 1b.[50] Both types 1a and 1b underwent massive expansions in their effective population size between 1940 and 1960. The expansion of HCV subtype 1b preceded that of subtype 1a by at least 16 years (95% credible interval: 15–17 years). Both types appear to have spread from the developed world to the developing world.
The genotype 2 strains from Africa can be divided into four clades that correlate with their country of origin: (1) Cameroon and Central African Republic (2) Benin, Ghana and Burkina Faso (3) Gambia, Guinea, Guinea-Bissau and Senegal (4) Madagascar.[51] There is also strong evidence now for the dissemination of hepatitis C virus genotype 2 from West Africa to the Caribbean by the Trans-Atlantic slave trade [52]
Genotype 3 is thought to have its origin in South East Asia.[53]
These dates from these various countries suggests that this virus may have evolved in South East Asia and was spread to West Africa by traders from Western Europe.[54] It was later introduced into Japan once that country's self-imposed isolation was lifted. Once introduced to a country its spread has been influenced by many local factors including blood transfusions, vaccination programmes, intravenous drug use and treatment regimes. Given the reduction in the rate of spread once screening for Hepatitis C in blood products was implemented in the 1990s it would seem that at least in recent times blood transfusion has been an important method of spreading for this virus. Additional work is required to determine the dates of evolution of the various genotypes and the timing of their spread across the globe.
9. Vaccination
Unlike hepatitis A and B, there is currently no vaccine to prevent hepatitis C infection.[55]
10. Current Research
The study of HCV has been hampered by the narrow host range of HCV.[56] The use of replicons has been successful but these have only been recently discovered.[57] HCV, as with most RNA viruses, exists as a viral quasispecies, making it very difficult to isolate a single strain or receptor type for study.[58][59]
Current research is focused on small-molecule inhibitors of the viral protease, RNA polymerase and other nonstructural genes. Two agents — Boceprevir by Merck[60] and Telaprevir by Vertex Pharmaceuticals Inc—both inhibitors of NS3 protease were approved for use on May 13, 2011 and May 23, 2011 respectively.
A possible association between low Vitamin D levels and a poor response to treatment has been reported.[61][62][63][64] In vitro work has shown that Vitamin D may be able to reduce viral replication.[65] While this work looks promising[66][67] the results of clinical trials are awaited.[68][69] However, it has been proposed that Vitamin D supplementation is important in addition to standard treatment, in order to enhance treatment response.[70]
Naringenin has been shown to block the assembly of intracellular infectious viral particles without affecting intracellular levels of the viral RNA or protein. Although the research is relatively new, naringenin may offer new insight into HCV therapeutic target.[70]
Other agents that are under investigation include nucleoside and nucleotide analogue inhibitors and non nucleoside inhibitors of the RNA dependent RNA polymerase, inhibitors of nonstructural protein 5A and host targeted compounds such as cyclophilin inhibitors and silibinin.[71]
Sofosbuvir for use against chronic hepatitis C infection was approved by the FDA December 6, 2013. It has been reported to be the first drug that has demonstrated safety and efficacy to treat certain types of HCV infection without the need for co-administration of interferon.[72] On November 22, the FDA approved simeprevir for use in combination with peginterferon-alfa and ribavirin.[73] Simeprevir has been approved in Japan for the treatment of chronic hepatitis C infection, genotype 1.[74]
There is also current experimental research on non drug related therapies. Oxymatrine, for example, is a root extract found in the continent of Asia that has been reported to have antiviral activity against HCV in cell cultures and animal studies. Small and promising human trials have shown no serious side effects and beneficial results, but they were too small to generalize conclusions.[70]
References
- "Characterization of a canine homolog of hepatitis C virus". Proc Natl Acad Sci U S A 108 (28): 11608–13. 2011. doi:10.1073/pnas.1101794108. PMID 21610165. Bibcode: 2011PNAS..10811608K. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3136326
- "Serology-enabled discovery of genetically diverse hepaciviruses in a new host". J. Virol. 86 (11): 6171–8. June 2012. doi:10.1128/JVI.00250-12. PMID 22491452. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3372197
- "Bats are a major natural reservoir for hepaciviruses and pegiviruses". Proc. Natl. Acad. Sci. U.S.A. 110 (20): 8194–9. May 2013. doi:10.1073/pnas.1303037110. PMID 23610427. Bibcode: 2013PNAS..110.8194Q. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3657805
- "Identification of rodent homologs of hepatitis C virus and pegiviruses". mBio 4 (2): e00216–13. 2013. doi:10.1128/mBio.00216-13. PMID 23572554. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3622934
- Dubuisson, Jean; Cosset, François-Loïc (2014). "Virology and cell biology of the hepatitis C virus life cycle – An update". Journal of Hepatology 61 (1): S3–S13. doi:10.1016/j.jhep.2014.06.031. PMID 25443344. https://dx.doi.org/10.1016%2Fj.jhep.2014.06.031
- Kaito, Masahiko; Ishida, Satoshi; Tanaka, Hideaki; Horiike, Shinichiro; Fujita, Naoki; Adachi, Yukihiko; Kohara, Michinori; Konishi, Masayoshi et al. (June 2006). "Morphology of hepatitis C and hepatitis B virus particles as detected by immunogold electron microscopy" (in en). Medical Molecular Morphology 39 (2): 63–71. doi:10.1007/s00795-006-0317-8. ISSN 1860-1480. PMID 16821143. https://dx.doi.org/10.1007%2Fs00795-006-0317-8
- "Topology of hepatitis C virus envelope glycoproteins". Rev. Med. Virol. 13 (4): 233–41. 2003. doi:10.1002/rmv.391. PMID 12820185. https://dx.doi.org/10.1002%2Frmv.391
- Castelli, Matteo; Clementi, Nicola; Pfaff, Jennifer; Sautto, Giuseppe A.; Diotti, Roberta A.; Burioni, Roberto; Doranz, Benjamin J.; Dal Peraro, Matteo et al. (2017-03-16). "A Biologically-validated HCV E1E2 Heterodimer Structural Model" (in En). Scientific Reports 7 (1): 214. doi:10.1038/s41598-017-00320-7. ISSN 2045-2322. PMID 28303031. Bibcode: 2017NatSR...7..214C. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=5428263
- Basu, Arnab; Beyene, Aster; Meyer, Keith; Ray, Ranjit (May 2004). "The Hypervariable Region 1 of the E2 Glycoprotein of Hepatitis C Virus Binds to Glycosaminoglycans, but This Binding Does Not Lead to Infection in a Pseudotype System". Journal of Virology 78 (9): 4478–4486. doi:10.1128/JVI.78.9.4478-4486.2004. ISSN 0022-538X. PMID 15078928. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=387685
- Kato N (2000). "Genome of human hepatitis C virus (HCV): gene organization, sequence diversity, and variation". Microb. Comp. Genom. 5 (3): 129–51. doi:10.1089/mcg.2000.5.129. PMID 11252351. https://dx.doi.org/10.1089%2Fmcg.2000.5.129
- Jubin R (2001). "Hepatitis C IRES: translating translation into a therapeutic target". Curr. Opin. Mol. Ther. 3 (3): 278–87. PMID 11497352. http://www.ncbi.nlm.nih.gov/pubmed/11497352
- "Crystal structure of the HCV IRES central domain reveals strategy for start-codon positioning". Structure 19 (10): 1456–66. October 2011. doi:10.1016/j.str.2011.08.002. PMID 22000514. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3209822
- Dubuisson J (2007). "Hepatitis C virus proteins". World J. Gastroenterol. 13 (17): 2406–15. doi:10.3748/wjg.v13.i17.2406. PMID 17552023. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4146758
- De Francesco R (1999). "Molecular virology of the hepatitis C virus". J Hepatol 31 (Suppl 1): 47–53. doi:10.1016/S0168-8278(99)80374-2. PMID 10622560. https://dx.doi.org/10.1016%2FS0168-8278%2899%2980374-2
- "Intrinsically unstructured domain 3 of hepatitis C Virus NS5A forms a "fuzzy complex" with VAPB-MSP domain which carries ALS-causing mutations". PLoS ONE 7 (6): e39261. 2012. doi:10.1371/journal.pone.0039261. PMID 22720086. Bibcode: 2012PLoSO...739261G. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3374797
- Jin, Z; Leveque, V; Ma, H; Johnson, K. A.; Klumpp, K (2012). "Assembly, purification, and pre-steady-state kinetic analysis of active RNA-dependent RNA polymerase elongation complex". Journal of Biological Chemistry 287 (13): 10674–83. doi:10.1074/jbc.M111.325530. PMID 22303022. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3323022
- "Replication of hepatitis C virus". Nat. Rev. Microbiol. 5 (6): 453–63. June 2007. doi:10.1038/nrmicro1645. PMID 17487147. https://dx.doi.org/10.1038%2Fnrmicro1645
- "Ligand-induced changes in hepatitis C virus NS5B polymerase structure". Antiviral Res. 88 (2): 197–206. November 2010. doi:10.1016/j.antiviral.2010.08.014. PMID 20813137. https://dx.doi.org/10.1016%2Fj.antiviral.2010.08.014
- "Crystal structures of the RNA-dependent RNA polymerase genotype 2a of hepatitis C virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors". J. Biol. Chem. 280 (18): 18202–10. May 2005. doi:10.1074/jbc.M413410200. PMID 15746101. https://dx.doi.org/10.1074%2Fjbc.M413410200
- "Substrate complexes of hepatitis C virus RNA polymerase (HC-J4): structural evidence for nucleotide import and de-novo initiation". J. Mol. Biol. 326 (4): 1025–35. February 2003. doi:10.1016/s0022-2836(02)01439-0. PMID 12589751. https://dx.doi.org/10.1016%2Fs0022-2836%2802%2901439-0
- "Non-nucleoside inhibitors binding to hepatitis C virus NS5B polymerase reveal a novel mechanism of inhibition". J. Mol. Biol. 361 (1): 33–45. August 2006. doi:10.1016/j.jmb.2006.05.074. PMID 16828488. https://dx.doi.org/10.1016%2Fj.jmb.2006.05.074
- "Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame". RNA 7 (5): 710–721. 2001. doi:10.1017/S1355838201010111. PMID 11350035. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1370123
- "Expression and characterization of Escherichia coli derived hepatitis C virus ARFP/F protein". Mol Biol (Mosk) 46 (2): 251–9. 2012. doi:10.1134/S0026893312020033. PMID 22670521. https://dx.doi.org/10.1134%2FS0026893312020033
- "Replication of hepatitis C virus". J. Gen. Virol. 81 (Pt 7): 1631–48. July 2000. doi:10.1099/0022-1317-81-7-1631. PMID 10859368. http://vir.sgmjournals.org/cgi/content/full/81/7/1631.
- Zeisel, M.; Barth, H.; Schuster, C.; Baumert, T. (2009). "Hepatitis C virus entry: molecular mechanisms and targets for antiviral therapy". Frontiers in Bioscience 14 (8): 3274–3285. doi:10.1016/j.cnsns.2008.11.006. PMID 19273272. Bibcode: 2009CNSNS..14.3274H. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3235086
- Kohaar, I.; Ploss, A.; Korol, E.; Mu, K.; Schoggins, J.; O'Brien, T.; Rice, C.; Prokunina-Olsson, L. (2010). "Splicing diversity of the human OCLN gene and its biological significance for hepatitis C virus entry". Journal of Virology 84 (14): 6987–6994. doi:10.1128/JVI.00196-10. PMID 20463075. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2898237
- "Unravelling hepatitis C virus replication from genome to function". Nature 436 (7053): 933–8. 2005. doi:10.1038/nature04077. PMID 16107832. Bibcode: 2005Natur.436..933L. https://dx.doi.org/10.1038%2Fnature04077
- Branch, A. D.; Stump, D. D.; Gutierrez, J. A.; Eng, F.; Walewski, J. L. (2005). "The Hepatitis C Virus Alternate Reading Frame (ARF) and Its Family of Novel Products: The Alternate Reading Frame Protein/F-Protein, the Double-Frameshift Protein, and Others". Seminars in Liver Disease 25 (1): 105–117. doi:10.1055/s-2005-864786. PMID 15732002. https://dx.doi.org/10.1055%2Fs-2005-864786
- "Interaction of hepatitis C virus proteins with host cell membranes and lipids". Trends Cell Biol 12 (11): 517–523. 2002. doi:10.1016/S0962-8924(02)02383-8. PMID 12446113. https://dx.doi.org/10.1016%2FS0962-8924%2802%2902383-8
- "Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex". J Virol 76 (12): 5974–84. 2002. doi:10.1128/JVI.76.12.5974-5984.2002. PMID 12021330. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=136238
- "Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner". Traffic 9 (8): 1268–82. 2008. doi:10.1111/j.1600-0854.2008.00767.x. PMID 18489704. https://dx.doi.org/10.1111%2Fj.1600-0854.2008.00767.x
- "Hepatitis C virus hijacks host lipid metabolism". Trends Endocrinol Metab 21 (1): 33–40. 2010. doi:10.1016/j.tem.2009.07.005. PMID 19854061. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2818172
- "Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS-5 region". J. Gen. Virol. 74 (Pt 11): 2391–9. November 1993. doi:10.1099/0022-1317-74-11-2391. PMID 8245854. http://vir.sgmjournals.org/cgi/pmidlookup?view=long&pmid=8245854.
- Nakano, Tatsunori; Lau, Gillian M. G.; Lau, Grace M. L.; Sugiyama, Masaya; Mizokami, Masashi (9 October 2011). "An updated analysis of hepatitis C virus genotypes and subtypes based on the complete coding region". Liver International 32 (2): 339–45. doi:10.1111/j.1478-3231.2011.02684.x. PMID 22142261. https://dx.doi.org/10.1111%2Fj.1478-3231.2011.02684.x
- "New hepatitis C virus (HCV) genotyping system that allows for identification of HCV genotypes 1a, 1b, 2a, 2b, 3a, 3b, 4, 5a, and 6a". J Clin Microbiol 35 (1): 201–7. 2007. PMID 8968908. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=229539
- Simmonds P; Bukh J; Combet C; Deléage G; Enomoto N; Feinstone S; Halfon P; Inchauspé G et al. (2005). "Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes". Hepatology 42 (4): 962–73. doi:10.1002/hep.20819. PMID 16149085. https://dx.doi.org/10.1002%2Fhep.20819
- "Treatment of chronic hepatitis C in Asia: when East meets West". J Gastroenterol Hepatol 24 (3): 336–345. 2009. doi:10.1111/j.1440-1746.2009.05789.x. PMID 19335784. https://dx.doi.org/10.1111%2Fj.1440-1746.2009.05789.x
- Muir, AJ; Bornstein, JD; Killenberg, PG; Atlantic Coast Hepatitis Treatment Group (2004). "Peginterferon alfa-2b and ribavirin for the treatment of chronic hepatitis C in blacks and non-Hispanic whites.". N Engl J Med 350 (22): 2265–71. doi:10.1056/NEJMoa032502. PMID 15163776. Erratum: doi:10.1056/nejm200409163511229 https://doi.org/10.1056%2Fnejm200409163511229
- Ge, DExpression error: Unrecognized word "et". (2009). "Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance". Nature 461 (7262): 399–401. doi:10.1038/nature08309. PMID 19684573. Bibcode: 2009Natur.461..399G. https://dx.doi.org/10.1038%2Fnature08309
- Rose, R; Markov, PV; Lam, TT; Pybus, OG (2013). "Viral evolution explains the associations among hepatitis C virus genotype, clinical outcomes, and human genetic variation". Infect Genet Evol 20: 418–21. doi:10.1016/j.meegid.2013.09.029. PMID 24140473. https://dx.doi.org/10.1016%2Fj.meegid.2013.09.029
- "Exposure of hepatitis C virus (HCV) RNA-positive recipients to HCV RNA-positive blood donors results in rapid predominance of a single donor strain and exclusion and/or suppression of the recipient strain". Journal of Virology 75 (5): 2059–66. 2001. doi:10.1128/JVI.75.5.2059-2066.2001. PMID 11160710. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=114790
- Shepard, CW; Finelli, L; Alter, MJ (Sep 2005). "Global epidemiology of hepatitis C virus infection". Lancet Infect Dis 5 (9): 558–67. doi:10.1016/S1473-3099(05)70216-4. PMID 16122679. http://www.thelancet.com/journals/laninf/article/PIIS1473-3099%2805%2970216-4/fulltext.
- Alter, MJ (Nov 2011). "HCV routes of transmission: what goes around comes around". Semin Liver Dis 31 (4): 340–6. doi:10.1055/s-0031-1297923. PMID 22189974. https://dx.doi.org/10.1055%2Fs-0031-1297923
- Pybus, OG; Markov, PV; Wu, A; Tatem, AJ (Jul 2007). "Investigating the endemic transmission of the hepatitis C virus". Int J Parasitol 37 (8–9): 839–49. doi:10.1016/j.ijpara.2007.04.009. PMID 17521655. https://dx.doi.org/10.1016%2Fj.ijpara.2007.04.009
- "Hepatitis C virus evolutionary patterns studied through analysis of full-genome sequences". J Mol Evol 54 (1): 62–70. 2002. doi:10.1007/s00239-001-0018-9. PMID 11734899. Bibcode: 2002JMolE..54...62S. https://dx.doi.org/10.1007%2Fs00239-001-0018-9
- "NS4A protein as a marker of HCV history suggests that different HCV genotypes originally evolved from genotype 1b". Virol. J. 8: 317. 2011. doi:10.1186/1743-422X-8-317. PMID 21696641. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3145594
- "Genetic history of hepatitis C virus in East Asia". J Virol 83 (2): 1071–82. 2009. doi:10.1128/JVI.01501-08. PMID 18971279. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2612398
- Kato N, Ueda Y, Sejima H, Gu W, Satoh S, Dansako H, Ikeda M, Shimotohno K (2019) Study of multiple genetic variations caused by persistent hepatitis C virus replication in long-term cell culture. Arch Virol
- "Investigation of the pattern of diversity of hepatitis C virus in relation to times of transmission". J Viral Hepat 4 (Suppl 1): 69–74. 1997. doi:10.1111/j.1365-2893.1997.tb00163.x. PMID 9097281. https://dx.doi.org/10.1111%2Fj.1365-2893.1997.tb00163.x
- "The global spread of hepatitis C virus 1a and 1b: a phylodynamic and phylogeographic analysis". PLoS Med. 6 (12): e1000198. December 2009. doi:10.1371/journal.pmed.1000198. PMID 20041120. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2795363
- "Phylogeography and molecular epidemiology of hepatitis C virus genotype 2 in Africa". J. Gen. Virol. 90 (Pt 9): 2086–96. September 2009. doi:10.1099/vir.0.011569-0. PMID 19474244. http://vir.sgmjournals.org/cgi/pmidlookup?view=long&pmid=19474244.
- Markov, PV et al. (2012). "Colonial History and Contemporary Transmission Shape the Genetic Diversity of Hepatitis C Virus Genotype 2 in Amsterdam". J Virol 86 (14): 7677–7687. doi:10.1128/JVI.06910-11. PMID 22573865. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3416291
- "Genetic diversity and evolution of hepatitis C virus—15 years on". J. Gen. Virol. 85 (Pt 11): 3173–88. November 2004. doi:10.1099/vir.0.80401-0. PMID 15483230. http://vir.sgmjournals.org/cgi/pmidlookup?view=long&pmid=15483230.
- "Reconstructing the origins of human hepatitis viruses". Philos Trans R Soc Lond B Biol Sci 356 (1411): 1013–26. 2001. doi:10.1098/rstb.2001.0890. PMID 11516379. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1088496
- "A new insight into hepatitis C vaccine development". J. Biomed. Biotechnol. 2010: 1–12. 2010. doi:10.1155/2010/548280. PMID 20625493. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2896694
- Rauch, A.; Gaudieri, S.; Thio, C.; Bochud, P. Y. (2009). "Host genetic determinants of spontaneous hepatitis C clearance". Pharmacogenomics 10 (11): 1819–1837. doi:10.2217/pgs.09.121. PMID 19891557. https://dx.doi.org/10.2217%2Fpgs.09.121
- "Hepatitis C virus virology and new treatment targets". Expert Rev Anti Infect Ther 7 (3): 329–50. April 2009. doi:10.1586/eri.09.12. PMID 19344246. https://dx.doi.org/10.1586%2Feri.09.12
- "The way forward in HCV treatment—finding the right path". Nat Rev Drug Discov 6 (12): 991–1000. December 2007. doi:10.1038/nrd2411. PMID 18049473. https://dx.doi.org/10.1038%2Fnrd2411
- Ahmed, Ali Mahmoud; Doheim, Mohamed Fahmy; Mattar, Omar Mohamed; Sherif, Nourin Ali; Truong, Duy Hieu; Pham T.L., Hoa; Hirayama, Kenji; Huy, Nguyen Tien (May 2018). "Beclabuvir in combination with asunaprevir and daclatasvir for hepatitis C virus genotype 1 infection: A systematic review and meta-analysis". J Med Virol 90 (5): 907–918. doi:10.1002/jmv.24947. PMID 28892235. https://dx.doi.org/10.1002%2Fjmv.24947
- "FDA approves Victrelis for Hepatitis C" (press release). FDA. May 13, 2011. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm255390.htm.
- "Classical and emerging roles of vitamin d in hepatitis C virus infection". Semin Liver Dis 31 (4): 387–398. 2011. doi:10.1055/s-0031-1297927. PMID 22189978. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=4107414
- "Vitamin D deficiency and a CYP27B1-1260 promoter polymorphism are associated with chronic hepatitis C and poor response to interferon-alfa based therapy". J Hepatol 54 (5): 887–893. 2011. doi:10.1016/j.jhep.2010.08.036. PMID 21145801. https://zenodo.org/record/3419700.
- "The vitamin D receptor gene bAt (CCA) haplotype impairs the response to pegylated-interferon/ribavirin-based therapy in chronic hepatitis C patients". Antivir. Ther. (Lond.) 17 (3): 541–7. 2012. doi:10.3851/IMP2018. PMID 22300961. https://dx.doi.org/10.3851%2FIMP2018
- "Complementary role of vitamin D deficiency and the interleukin-28B rs12979860 C/T polymorphism in predicting antiviral response in chronic hepatitis C". Hepatology 53 (4): 1118–26. 2011. doi:10.1002/hep.24201. PMID 21480318. https://dx.doi.org/10.1002%2Fhep.24201
- "Vitamin D: an innate antiviral agent suppressing hepatitis C virus in human hepatocytes". Hepatology 54 (5): 1570–9. 2011. doi:10.1002/hep.24575. PMID 21793032. https://dx.doi.org/10.1002%2Fhep.24575
- "Vitamin D supplementation improves sustained virologic response in chronic hepatitis C (genotype 1)-naïve patients". World J Gastroenterol 17 (47): 5184–90. 2011. doi:10.3748/wjg.v17.i47.5184. PMID 22215943. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3243885
- "Vitamin D supplementation improves response to antiviral treatment for recurrent hepatitis C". Transpl Int 24 (1): 43–50. 2011. doi:10.1111/j.1432-2277.2010.01141.x. PMID 20649944. https://dx.doi.org/10.1111%2Fj.1432-2277.2010.01141.x
- "Review article: the extra-skeletal effects of vitamin D in chronic hepatitis C infection". Aliment. Pharmacol. Ther. 35 (6): 634–46. March 2012. doi:10.1111/j.1365-2036.2012.05000.x. PMID 22316435. https://dx.doi.org/10.1111%2Fj.1365-2036.2012.05000.x
- "Diagnostic and therapeutical role of vitamin D in chronic hepatitis C virus infection". Front Biosci 1 (4): 1276–1286. 2012. doi:10.2741/e458. https://dx.doi.org/10.2741%2Fe458
- Halegoua-De Marzio, Dina; Fenkel, Jonathan (January 27, 2014). "Alternative medications in Hepatitis C infection". World Journal of Hepatology 6 (1): 9–16. doi:10.4254/wjh.v6.i1.9. PMID 24653790. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=3953807
- "Antiviral strategies in hepatitis C virus infection". J. Hepatol. 56 (Suppl 1): S88–100. 2012. doi:10.1016/S0168-8278(12)60010-5. PMID 22300469. https://dx.doi.org/10.1016%2FS0168-8278%2812%2960010-5
- Press announcement, FDA, December 6 2013 https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm377888.htm
- "FDA approves new treatment for hepatitis C virus". Food and Drug Administration. Nov 22, 2013. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm376449.htm.
- "Medivir: Simeprevir has been approved in Japan for the treatment of genotype 1 chronic hepatitis C infection". The Wall Street Journal. September 27, 2013. https://www.wsj.com/article/PR-CO-20130927-900120.html.