Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Zhao-Qing Shen.
An age-dependent decrease in CDGSH (Cys-Asp-Gly-Ser-His) iron-sulfur domain 2 (CISD2) expression during the natural aging of mice has been reported in a range of tissues, including the brain, spinal cord, skeletal muscle, heart, and skin. However, the rate of CISD2 downregulation varies from tissue to tissue.
- CISD2
- aging
- rejuvenation
- longevity
2.1. CISD2 Is One of a Limited Number of Pro-Longevity Genes in Mammals
1. CISD2 Is One of a Limited Number of Pro-Longevity Genes in Mammals
CDGSH (Cys-Asp-Gly-Ser-His) iron-sulfur domain 2 (CISD2) deficiency is associated with many hallmarks of aging, including mitochondrial dysfunction accompanied by autophagy and cell death, disorganized proteostasis, deregulated nutrient sensing with metabolism disturbances, stem cell exhaustion, and alterations in intercellular communication [39][1]. The earliest manifestation of mitochondrial dysfunction appears in the organs with the highest energy demand, namely the heart, skeletal muscle, and nervous system. Furthermore, CISD2 knockout (CISD2 KO) mice display a panel of phenotypic features that are indicative of premature aging; these include a shortened lifespan accompanied by multiple age-associated organ dysfunctions (cardiac electromechanical dysfunction, sarcopenia, and degeneration of the nervous system), as well as dysregulation of whole body energy metabolism. Notably, many of these manifestations are consistent with the symptoms present in Wolfram syndrome 2 (WFS2) patients. Moreover, in CISD2 transgenic (CISD2 TG) mice, ourne previous studies revealed that a persistently high level of CISD2 extended their median and maximum lifespan without any apparent deleterious side effects [14][2]. A persistently high level of CISD2 expression ameliorated age-associated degeneration of the skin, skeletal muscles, neurons, and cardiac performance during old age [14,17,18,40][2][3][4][5].
2.2. CISD2 in Cardiac Aging
2. CISD2 in Cardiac Aging
A major risk factor for cardiovascular disease is aging. Thus, it is critical to preserve cardiac functioning during aging [41][6]. One of our previous studies showed that CISD2 plays an essential role in maintaining normal cardiac structure and function [18][4]. The level of cardiac CISD2 present in naturally aged mice is significantly reduced by approximately 50% compared with that found in 3-month old (3-mo) young mice (Figure 1A). Loss of CISD2 in CISD2 KO mice resulted in an aged phenotype of the heart, including perivascular fibrosis, deterioration of the intercalated discs (ICDs), disorganized myofibrils, and degenerated and swollen mitochondria. Subsequently, these changes brought about myocardial degeneration and impairment of the electromechanical performance of CISD2 KO mice at a young age (3-mo). Maintenance of a high level of CISD2 protein in the heart is crucial to preserving the integrity of the ICDs, which allows for synchronous contraction and relaxation of cardiomyocytes as a single functional organ. ICD degeneration, including lateralization of gap junctions, maldistribution of desmosomes, and a breakdown of the fascia adherens, appears in CISD2 KO mice at a young age, due to the lack of CISD2, and in naturally aged mice, due to the decrease in CISD2 with aging. ICD degeneration leads to electromechanical impairment. Loss of CISD2 also results in derangement of mitochondrial ultrastructure and the abnormal functioning of oxidative phosphorylation.
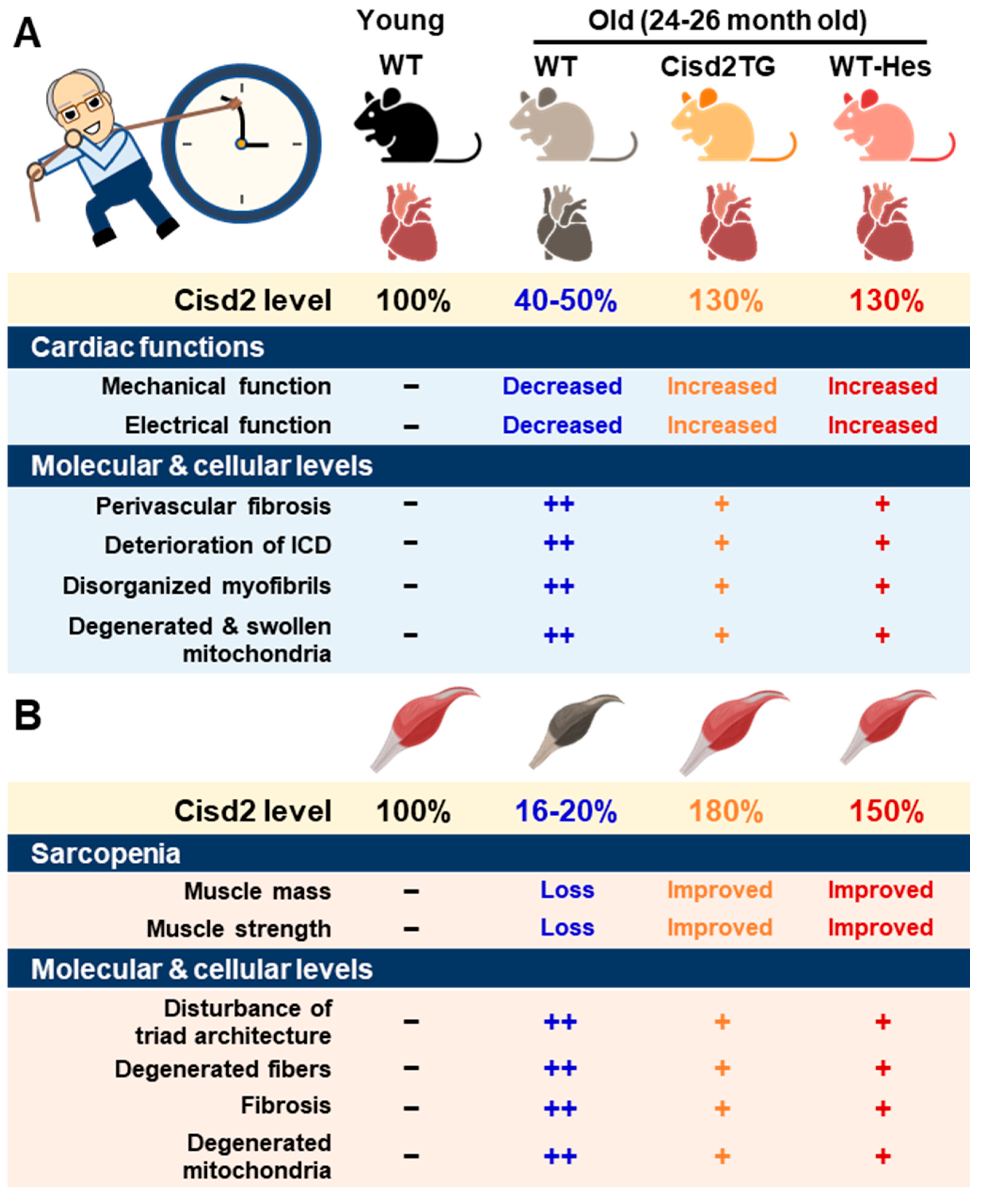 On the other hand, a persistently high level of CISD2 during old age, which was achieved by a transgenic approach in the CISD2 TG mice, appears to preserve cardiac functions during aging [18,42][4][7]. Intriguingly, cardiac-specific induction of CISD2 overexpression at 18 mos of age appeared to retard cardiac aging and reverse age-associated cardiac dysfunction to a measurable extent in mice. Enhanced expression of CISD2 at a late-life stage is thus able to attenuate the age-associated decline in mitochondrial membrane potentials and respiratory function, as well as minimize ROS production, thereby rejuvenating an aging heart [42][7].
On the other hand, a persistently high level of CISD2 during old age, which was achieved by a transgenic approach in the CISD2 TG mice, appears to preserve cardiac functions during aging [18,42][4][7]. Intriguingly, cardiac-specific induction of CISD2 overexpression at 18 mos of age appeared to retard cardiac aging and reverse age-associated cardiac dysfunction to a measurable extent in mice. Enhanced expression of CISD2 at a late-life stage is thus able to attenuate the age-associated decline in mitochondrial membrane potentials and respiratory function, as well as minimize ROS production, thereby rejuvenating an aging heart [42][7].
Figure 1. CISD2 attenuates heart and skeletal muscle aging. (A) An age-dependent decrease in CISD2 correlates with cardiac dysfunctions and molecular pathological alterations. Whole-body CISD2 overexpression (CISD2 TG) delays age-associated cardiac damage. In addition, the CISD2 activator, hesperetin (Hes), reverses age-associated cardiac damage via the activation of CISD2. (B) Age-dependent CISD2 reduction leads to sarcopenia and pathological alterations. Maintaining a high level of CISD2 by transgenic overexpression, or by hesperetin treatment, is able to protect skeletal muscle from age-related pathological and molecular damage. The figure was created with BioRender.com.
2.3. CISD2 in Muscle Aging
3. CISD2 in Muscle Aging
One of the most prominent features during aging is the loss of skeletal muscle mass and strength/function, which is referred to as sarcopenia [43][8]. The proteomic signature of sarcopenia reveals that mitochondrial dysfunction and alterations in intracellular Ca2+ homeostasis are significantly associated with sarcopenia [44,45][9][10]. The preferential loss of muscle during aging involves the glycolytic fast-twitch fibers of the gastrocnemius muscle, in particular [46,47][11][12]. There was a significant downregulation of CISD2 protein in skeletal muscle, specifically the gastrocnemius, of WT mice at middle age (at 13 mo; only ~30% of CISD2 protein remained) and old age (at 24 mo; only 16 to 20% of CISD2 protein remained), which suggests that CISD2 may play a vital role in skeletal muscle aging (Figure 1B) [17,20][3][13]. Notably, mice carrying the muscle-specific knockout of CISD2 (CISD2 mKO) at a young age (3 mo) showed similar phenotypic characteristics to those observed in naturally aged mice; this includes degeneration of skeletal muscles, which is accompanied by impairment of proteostasis and destruction of mitochondria and the ER/SR. In these CISD2 mKO mice, CISD2 deficiency induced endoplasmic reticulum (ER) stress and unfolded protein response (UPR) signaling in the muscle at a young age (3 mo). In addition, CISD2 deficiency led to alterations in the antioxidant defense pathways of both young CISD2 mKO at 3 mo and naturally aged WT mice at 24–26 mo. Subsequently, the resulting elevated ROS levels appeared to increase the oxidative modifications of SERCA1 (sarco/endoplasmic reticulum Ca2+-ATPase (SERCA)), thereby damaging enzymatic activity of this enzyme, which brought about an impairment of Ca2+ re-uptake into the ER/SR. This reduction in the Ca2+ level of the ER/SR appeared to compromise the functioning of many chaperones, which are proteins that assist others to fold properly; this folding is Ca2+-dependent. The end result was that these abnormalities formed a vicious cycle that was exacerbated during aging. Intriguingly, in CISD2 TG mice, which are a long-lived mouse model, a persistently high level of CISD2 expression was able to protect skeletal muscles from age-dependent mass loss and functional decline [14][2]. Transmission electron microscopy (TEM) examination further revealed that CISD2 appears to ameliorate the age-associated ultrastructural abnormalities normally present in the skeletal muscles, namely a disturbance of triad structure, disorganization of fibers, and degeneration of mitochondria [14][2].2.4. CISD2 in Liver Aging
4. CISD2 in Liver Aging
The liver is one of the most important metabolic organs for maintaining whole body health and plays a pivotal role in several biological processes, such as lipid metabolism, glucose homeostasis, xenobiotic and drug detoxification, as well as in the biosynthesis and secretion of plasma proteins. Accordingly, age-associated liver dysfunction not only affects the liver per se, but also contributes to the development of many other age-related diseases, such as various cardiovascular diseases, metabolic syndrome, diabetes, and a number of cancers. During liver aging, multiple age-related alterations, including pathological changes, functional decline, and metabolic dysregulation have been found [48][14]. Regarding the hallmarks of aging in the liver, there are several remarkable changes; these include a loss of proteostasis, the induction of ER stress, presence of dysfunctional mitochondria, increase in oxidative stress, and presence of cellular senescence. In particular, the mitochondria appear to play a crucial role in the aging of the liver [48,49][14][15]. Previously, wone group demonstrated that CISD2 maintained cellular homeostasis via the modulation of mitochondrial function, ER integrity, redox status, and intracellular Ca2+ homeostasis [20][13]. Intriguingly, in the aging liver, the level of CISD2 protein has been found to decrease to about 50% in old mice at 26 mo compared with young mice at 3 mo [50][16]. This result suggests that an age-associated decline in CISD2 expression seems to be involved in the liver aging process. Importantly, in Cisd2 TG mice at 26 mo, a persistently high level of CISD2 was shown to attenuate age-related liver pathology, reduce oxidative stress, and preserve a youthful pattern of gene expression, as revealed by transcriptomic and proteomic analyses. Such an effect thereby maintains normal metabolic function within the liver during aging [40,50][5][16]. Mechanistically, CISD2 seems to suppress the age-related dysregulation of various transcription mediators, such as Nrf2, IL-6, and Hnf4a; this, in turn, may help to preserve the transcriptional network, thereby resulting in a younger profile of gene expression in the liver. Interestingly, CISD2 appears to function in a cell autonomous manner when protecting hepatocytes from age-associated damage. In the AML12-CISD2 KO hepatocyte cell line, CISD2 deficiency resulted in lipid accumulation, mitochondrial dysfunction, and increased oxidative stress; additionally, CISD2 deficiency also led to dysregulation of the downstream target genes of Nrf2 and Hnf4a. On the other hand, in the AML12-CISD2 RE hepatocyte cell line, in which CISD2 was re-expressed in the CISD2 KO hepatocytes, all of the cellular phenotypes disappeared [50][16]. Indeed, CISD2 haploinsufficiency disrupts calcium homeostasis, induces ER stress, and promotes hepatocellular carcinoma (HCC). In contrast, CISD2 overexpression prevents HCC development and protects against HBx-mediated liver damage and lipotoxicity [29][17]. Taken together, genetic evidence reveals that downregulation of CISD2 accelerates liver aging and promotes the development of age-related liver diseases, including NAFLD, NASH, and HCC. Conversely, a high level of CISD2 protects the liver from oxidative stress, preserves mitochondrial functioning, and attenuates the age-related loss of proteostasis, as well as maintains a youthful pattern of gene expression [40,50][5][16].References
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- Wu, C.Y.; Chen, Y.F.; Wang, C.H.; Kao, C.H.; Zhuang, H.W.; Chen, C.C.; Chen, L.K.; Kirby, R.; Wei, Y.H.; Tsai, S.F.; et al. A persistent level of Cisd2 extends healthy lifespan and delays aging in mice. Hum. Mol. Genet. 2012, 21, 3956–3968.
- Huang, Y.L.; Shen, Z.Q.; Wu, C.Y.; Teng, Y.C.; Liao, C.C.; Kao, C.H.; Chen, L.K.; Lin, C.H.; Tsai, T.F. Comparative proteomic profiling reveals a role for Cisd2 in skeletal muscle aging. Aging Cell 2018, 17, e12705.
- Yeh, C.H.; Shen, Z.Q.; Hsiung, S.Y.; Wu, P.C.; Teng, Y.C.; Chou, Y.J.; Fang, S.W.; Chen, C.F.; Yan, Y.T.; Kao, L.S.; et al. Cisd2 is essential to delaying cardiac aging and to maintaining heart functions. PLoS Biol. 2019, 17, e3000508.
- Huang, C.H.; Huang, Y.L.; Shen, Z.Q.; Lin, C.H.; Tsai, T.F. Cisd2 preserves the youthful pattern of the liver proteome during natural aging of mice. Biomedicines 2021, 9, 1229.
- Pagan, L.U.; Gomes, M.J.; Gatto, M.; Mota, G.A.F.; Okoshi, K.; Okoshi, M.P. The role of oxidative stress in the aging heart. Antioxidants 2022, 11, 336.
- Yeh, C.H.; Chou, Y.J.; Chu, T.K.; Tsai, T.F. Rejuvenating the aging heart by enhancing the expression of the Cisd2 prolongevity Gene. Int. J. Mol. Sci. 2021, 22, 11487.
- Marzetti, E. Musculoskeletal aging and sarcopenia in the elderly. Int. J. Mol. Sci. 2022, 23, 2808.
- Protasi, F.; Pietrangelo, L.; Boncompagni, S. Improper remodeling of organelles deputed to Ca(2+) handling and aerobic ATP production underlies muscle dysfunction in ageing. Int. J. Mol. Sci. 2021, 22, 6195.
- Zampino, M.; Tanaka, T.; Ubaida-Mohien, C.; Fantoni, G.; Candia, J.; Semba, R.D.; Ferrucci, L. A plasma proteomic signature of skeletal muscle mitochondrial function. Int. J. Mol. Sci. 2020, 21, 9540.
- Lexell, J. Human aging, muscle mass, and fiber type composition. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, 11–16.
- Sinha-Hikim, I.; Sinha-Hikim, A.P.; Parveen, M.; Shen, R.; Goswami, R.; Tran, P.; Crum, A.; Norris, K.C. Long-term supplementation with a cystine-based antioxidant delays loss of muscle mass in aging. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 749–759.
- Shen, Z.Q.; Huang, Y.L.; Teng, Y.C.; Wang, T.W.; Kao, C.H.; Yeh, C.H.; Tsai, T.F. CISD2 maintains cellular homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118954.
- Hunt, N.J.; Kang, S.W.S.; Lockwood, G.P.; Le Couteur, D.G.; Cogger, V.C. Hallmarks of aging in the liver. Comput. Struct. Biotechnol. J. 2019, 17, 1151–1161.
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258.
- Huang, Y.L.; Shen, Z.Q.; Huang, C.H.; Lin, C.H.; Tsai, T.F. Cisd2 slows down liver aging and attenuates age-related metabolic dysfunction in male mice. Aging Cell 2021, 20, e13523.
- Shen, Z.Q.; Chen, Y.F.; Chen, J.R.; Jou, Y.S.; Wu, P.C.; Kao, C.H.; Wang, C.H.; Huang, Y.L.; Chen, C.F.; Huang, T.S.; et al. CISD2 haploinsufficiency disrupts calcium homeostasis, causes nonalcoholic fatty liver disease, and promotes hepatocellular carcinoma. Cell Rep. 2017, 21, 2198–2211.
More