Heart failure (HF) is a major public health problem worldwide, presented by an inability of the heart to provide metabolic demands and perfusion of organs/tissues and characterized by symptoms and signs, including shortness of breath, fatigue, rapid to irregular heart rate, lung crepitations, elevated jugular venous pressure, and peripheral tissue edema
[1][2][1,2]. HF affects more than 26 million adults worldwide and an estimated 6 million American adults have HF
[2][3][2,3], in which coronary heart disease (myocardial infarction, MI)-induced HF with reduced ejection fraction (HFrEF) accounts for about 50% of all HF cases
[4][5][6][7][4,5,6,7]. Considering the stable incidence of HF with an annual increase, the actual burden of treatment and diagnosis in patients with HF has obviously exceeded the projected burden in the United States and worldwide, especially accounting for other factors, including an increased comorbidity burden and advancing age of the population
[2][8][9][2,8,9]. Despite advances in diagnosis and therapeutic management of HF, HF still has a high morbidity and mortality rate. As a major feature of HF, cardiac sympathetic overactivation
[10][11][12][13][14][10,11,12,13,14] triggers malignant arrhythmias and sudden cardiac death
[15][16][17][18][19][20][21][22][23][15,16,17,18,19,20,21,22,23], which accounts for nearly 50–60% of the mortality in HF patients
[20][24][25][26][27][28][29][30][31][32][33][34][35][20,24,25,26,27,28,29,30,31,32,33,34,35]. The role of cardiac sympathetic hyperactivation in HF is highlighted by the use of β-blockers and cardiac sympathetic denervation as the key approach to the current therapy of HF
[36][37][38][39][40][41][42][43][36,37,38,39,40,41,42,43]. However, such pharmacological treatment may not be ideal because some studies have demonstrated that β-blockers do not provide satisfactory protection against sudden cardiac death, and some patients are either intolerant or refractory to this therapy
[44][45][46][47][48][49][50][44,45,46,47,48,49,50]. Additionally, despite being an alternative in managing refractory ventricular arrhythmias
[38][43][51][52][38,43,51,52], cardiac sympathetic denervation has adverse complications (including Horner’s syndrome, hyperhidrosis, paresthesia, and sympathetic fight/fight response loss) that severely limit the use of procedures in HF patients
[53][54][53,54]. These drawbacks have increased the focus on exploring the mechanisms responsible for HF-increased cardiac sympathetic activation and on identifying effective therapeutic interventions, which are crucial for improving prognosis of HF and reducing its mortality.
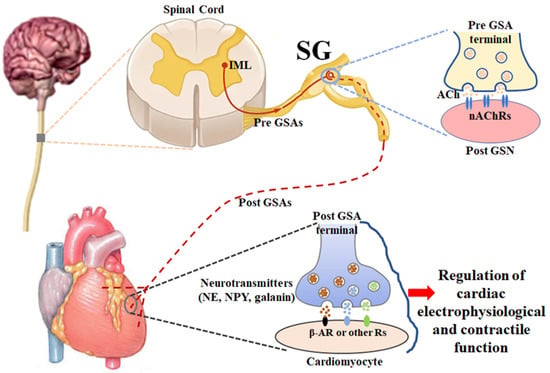
2. Anatomy and Physiology of Stellate Ganglia (Figure 1)
The sympathetic nervous system is one of the two divisions of the autonomic nervous system, the other being the parasympathetic nervous system. The sympathetic nervous system is composed of preganglionic and postganglionic neurons that are involved in signal transmission to regulate a variety of functions in all peripheral organs/tissues. The cardiac preganglionic sympathetic neurons originate in the intermediolateral column of the spinal cord in the thoracic region with the somata located in the gray rami communicantes bilaterally and symmetrically. Their axons are very short and pass through the white rami communicantes to form the synapses with cardiac postganglionic sympathetic neurons located in the lower cervical and upper thoracic paravertebral ganglia, releasing a neurotransmitter, acetylcholine, from cardiac preganglionic nerve terminals
[70][71][70,71]. Usually, the 7th cervical and 1st thoracic paravertebral sympathetic ganglia fuse into stellate ganglia, and the latter play a key role in a substantial amount of cardiac neurotransmission
[71][72][73][71,72,73]. When acetylcholine released from cardiac preganglionic sympathetic nerve endings activates nicotinic acetylcholine receptors on cardiac postganglionic sympathetic neurons in stellate ganglia, the longer cardiac postganglionic sympathetic nerve terminals innervated the heart release some neurotransmitters (such as norepinephrine, neuropeptide Y, and galanin) to regulate the functions of the heart through the activation of adrenergic receptors and other peptide receptors
[72][74][72,74].
Figure 1. A schematic diagram illustrating the anatomy and physiology of stellate ganglia. ACh: acetylcholine; β-AR: β-adrenergic receptor; IML: intermediolateral nucleus; nAChR: nicotinic acetylcholine receptor; NE: norepinephrine; NPY: neuropeptide Y; Pre GSA: preganglionic sympathetic axon; Post GSA: postganglionic sympathetic axon; Post GSN: postganglionic sympathetic neuron; SG: stellate ganglion.
In the physiological condition, the sympathetic nervous system is responsible for up/downregulating various homeostatic mechanisms in many organs/tissues, especially mediating the fight-or-flight response in situations in which survival is threatened
[75][76][75,76]. Norepinephrine, which is released from cardiac postganglionic sympathetic neurons in stellate ganglia with their nerve terminals, binds with beta-adrenergic receptors to affect cardiac electrophysiological and contractile functions, including heart rate, cardiac conduction, and myocardial contraction, which finally regulates cardiac output to supply the whole body with oxygenated blood and nutrients
[14][75][14,75]. It is widely recognized that cardiac postganglionic sympathetic nerve terminals innervate the sinoatrial node, atrioventricular node, His bundle, and contractile myocardium
[77]. However, the innervation of the heart with cardiac postganglionic sympathetic neurons in the stellate ganglia presents lateral and regional variations (such as anterior/posterior and left/right divisions of the heart)
[78]. In particular, there is an obvious variation and overlap in the innervation of the cardiac tissues from the left and right stellate ganglia
[78][79][78,79]. The sinoatrial node is primarily innervated by cardiac postganglionic sympathetic nerve terminals from the right stellate ganglion
[78]. The conduction system, including the sinoatrial node, atrioventricular node, and His bundle, is more densely innervated than the contractile myocardium
[80][81][80,81]. Compared to the endocardium of the heart, there is a high density of postganglionic sympathetic innervation on the epicardium of the heart
[78][81][78,81]. The posterior surface of the heart is mostly innervated by cardiac postganglionic sympathetic nerve terminals from the left stellate ganglion, whereas the anterior surface of the heart is principally innervated by sympathetic nerve terminals from the right stellate ganglion, measured by activation recovery internal (ARI) shortening as a probe of functional innervation
[78]. Additionally, it is possible that certain areas of the heart are not innervated by cardiac postganglionic sympathetic nerve terminals from both sides of the stellate ganglia.
3. Remodeling of Cardiac Postganglionic Sympathetic Neurons and Its Role in Cardiac Sympathetic Overactivation, Malignant Arrhythmias, and Cardiac Sudden Death in HF
Although cardiac sympathetic remodeling can contribute to cardiac sympathetic overactivation and has not been systematically explored during HF progression, scattered information about HF-triggered cardiac sympathetic remodeling is demonstrated by most previous studies, including
our
esearchers' work. These include structural and functional changes in cardiac postganglionic sympathetic cell somata and their nerve terminals.
3.1. Structural Remodeling in Cardiac Postganglionic Sympathetic Neurons Located in Stellate Ganglia
The majority of sympathetic nerves projecting to the heart originate in cardiac sympathetic postganglionic neurons located in stellate ganglia. There are limited data on the actual remodeling of cardiac sympathetic neuronal structures in stellate ganglia. One previous study demonstrated that stellate ganglionic nerve sprouting and density are elevated at one to four weeks after coronary artery ligation-induced rabbit myocardial infarction, which is mediated by nerve growth factor
[82][92]. In a porcine chronic myocardial infarction model (six weeks after left anterior coronary descending artery occlusion-induced myocardial infarction), chronic myocardial infarction significantly increased the size of neuronal somata in the left stellate ganglion
[83][93]. Using growth-associated protein 43, synaptophysin, and tyrosine hydroxylase as immunohistochemistry markers of synapses and sympathetic neurons in stellate ganglia, Han et al. found that the synaptic density and size of sympathetic neurons in the left stellate ganglion increased in dogs two months post-myocardial infarction
[84][94]. Tan et al. also reported that the sympathetic nerve density (immunoreactivity of tyrosine hydroxylase) in stellate ganglia was markedly increased in canines 12 weeks after premature ventricular contraction-induced cardiomyopathy
[85][95]. Similarly, Ajijola et al. demonstrated that the size of stellate ganglionic neurons increased with an increase in the neuronal adrenergic phenotype and neuropeptide Y-positive neurons in pigs at 6 weeks post left circumflex or right coronary artery occlusion-induced myocardial infarction
[86][96]. In humans with cardiomyopathy, the size of the stellate ganglionic neurons is significantly increased without ganglionic fibrosis and changes in the neuronal density (cell number/tissue area) and synaptic density
[87][97]. Although these studies in myocardial infarction-induced animal HF models and humans with cardiomyopathy are not totally consistent and it is unclear how structural alterations of cardiac postganglionic sympathetic neurons affect the progression and prognosis of HF, the morphological changes in these neurons could be associated with increased stellate ganglionic nerve activities and further related to cardiac sympathetic overactivation and malignant arrhythmias. Certainly, investigation of the structures of subcellular organelles (including nucleus, mitochondria, lysosome, and secretory vesicles) by electron microscopy is necessary to explore the cellular and molecular mechanisms underlying the structural remodeling of cardiac postganglionic sympathetic neuronal somata in HF.
3.2. Functional Remodeling in Cardiac Postganglionic Sympathetic Neurons Located in Stellate Ganglia
The function of neurons is to transmit electrical signals over long distances through the generation of action potentials. The left stellate ganglionic nerve activity is increased in ambulatory dogs with pacing- or coronary artery occlusion-induced HF
[84][88][94,98]. Tu et al. demonstrated that the cell excitability in cardiac postganglionic sympathetic neurons located in stellate ganglia increases in coronary artery ligation-induced HF rats
[89][99]. Although various types of ion channels (such as voltage-gated sodium, calcium, and potassium channels) can contribute to the generation of action potentials in cardiac postganglionic sympathetic neurons, voltage-gated calcium channels should be considered as the mechanism governing the increased cell excitability of these sympathetic neurons in HF because calcium influx through voltage-gated calcium channels is a key trigger for the release of neurotransmitters from neuronal nerve terminals
[90][91][92][100,101,102]. There are five subtypes of voltage-gated calcium channels (T, L, N, P/Q, and R) functionally characterized in central and peripheral neurons
[93][94][103,104]. A pore-forming α-subunit in all subtypes of calcium channels determines the biophysical and pharmacological properties of calcium channels
[95][105]. There are three major families of α-subunits: (1) Cav1 (Cav1.1, Cav1.2, and Cav1.3) encodes L-type calcium channels; (2) Cav2 encodes P/Q (Cav2.1), N (Cav2.2), and R (Cav2.3) types of calcium channels; and (3) Cav3 encodes T-type calcium channels
[95][96][105,106]. In fact, Tu et al. reported that the L, N, P/Q, and R types of calcium channels are expressed in cardiac postganglionic sympathetic neurons
[89][99]. However, HF only increases N-type calcium currents and does not affect the mRNA and protein expression of all calcium channel subtypes in these sympathetic neurons
[89][99]. Some previous studies demonstrated that N-type calcium channels, predominantly expressed in the nervous system, play an important role in modulating neurotransmitter release at sympathetic neve terminals
[97][98][107,108]. More importantly, increased N-type calcium currents in cardiac postganglionic sympathetic neurons contribute to the elevated cell excitability of these neurons, cardiac sympathetic overactivation, and malignant arrhythmias in HF
[99][100][109,110]. Until now, there has been no report about the involvement of other ion channels in HF-increased cell excitability of cardiac postganglionic sympathetic neurons.
3.3. Structural Remodeling in Cardiac Postganglionic Sympathetic Nerve Terminals
Cardiac sympathetic nerve terminals are directly embedded in the myocardium with a heterogeneous distribution. Most previous studies reported the information about structural remodeling in cardiac postganglionic sympathetic nerve terminals during HF progression from acute myocardial infarction to chronic HF, with no consistent conclusion. Acute myocardial infarction could result in sympathetic nerve terminal denervation in the scar and viable myocardium beyond the infarcted area
[101][102][103][111,112,113]. Then, the regeneration of sympathetic nerve terminals in the heart has been characterized by nerve spouting and a high density of nerve fibers in the periphery of the necrotic myocardium of failed hearts
[104][105][106][114,115,116]. Additionally, some previous studies also reported cardiac sympathetic nerve terminal denervation in HF
[107][108][117,118]. Regions of cardiac sympathetic nerve terminal denervation and hyperinnervation are present in the same failed heart to form the heterogeneity of the cardiac sympathetic nerve distribution
[109][110][119,120]. Iodine-123 meta-iodobenzylguanidine (
123I-MIBG) or other radiolabeled neurotransmitter analogs (including the recently used F-18 meta-fluorobenzylguanidine) and cardiac neurotransmission imaging with single-photon emission computed tomography (SPECT) and positron emission tomography (PET) have been employed to noninvasively assess the integrity of human NET and further evaluate cardiac sympathetic nerve innervation
[111][112][113][121,122,123]. However, poor imaging quality, difficulty in distinguishing different cardiac structures, and high cost limit this technique’s application in animal studies, especially small animal studies. Additionally, previous studies used immunohistochemical staining in several myocardial slices to evaluate the structural remodeling of cardiac sympathetic nerve terminals, which cannot represent the distribution of cardiac sympathetic nerve terminals in the whole heart with a neglected heterogeneous distribution of nerve terminals. Although these structural alterations of cardiac postganglionic sympathetic nerve terminals are considered to create a high-yield substrate for malignant arrhythmias in HF
[114][124], the conclusion from these previous studies should be questioned. Using three-dimensional assessment of the cardiac sympathetic network in cleared transparent murine hearts, one recent study demonstrated both cardiac sympathetic nerve terminal hyperinnervation and denervation in the whole heart at 2 weeks post myocardial infarction
[115][125]. It is not clear whether the same phenomenon (sympathetic nerve terminal hyperinnervation and denervation) is also present in the whole heart with HF using three-dimensional assessment of the cardiac sympathetic network. Therefore, the timing and patterns of the cardiac sympathetic nerve terminal remodeling in HF should be re-evaluated in future studies. Indeed, structural remodeling and norepinephrine release in cardiac postganglionic sympathetic nerve terminals in HF should be combined to assess the association of cardiac sympathetic activation and malignant arrhythmias because it is unclear whether reinnervated cardiac sympathetic nerve terminals can release norepinephrine like mature sympathetic nerve terminals in the heart.