1. Focus on LncRNAs
One of the types of non-coding RNAs that span higher than 200 nucleotides in length, possess no mechanism to code for proteins, and are seen to have a little expression in certain tissues are considered as long non-coding RNAs. Some lncRNAs may show exceptions to these criteria. It is quite evident from the recent research that the abnormal expression of LncRNAs plays a significant role in
cancer stem cells (CSCs
)’ metabolism. They regulate gene expression by the following approaches: as a modulator of gene expression; as a decoy to lead the transcription factor elsewhere from a target site; as a competitor to hinder the attachment of other molecules to the target site; as a chaperone for molecules to attach to a certain segment and as a scaffold that enhances the association of different proteins into different complexes
[1][79].
Different types of lncRNAs regulate the functions of CSCs by altering and modulating different transcription factors, enzymes, niches, and signaling pathways. LncRNA H19 is seen to maintain the stemness in glioma
[2][80] and BCSCs
[3][81]. Contrastingly, lncRNA ROR plays a repressive role in glioma SCs by negatively altering the expressions of stem cell factor KLF4
[4][82]. LncRNA CUDR leads to high H3K4 trimethylation by activating the associations between SET1A and pRB1in hepatocellular carcinoma (HCC)
[5][83]. It works with Cyclin-D to stimulate the self-renewal and proliferation of hepatocellular carcinoma stem cells by increasing the expression of telomerase reverse transcriptase (TERT) and C-Myc
[6][84]. In prostate cancer, the PCR2-LncRNA HOTAIR association represses the androgen receptor transcription by complexing to its 5′-flanking promoter region and enhancing prostate cancer stem/progenitor cells and invasion
[7][85]. In breast cancer, LncRNA HOTAIR leads to the induction of
epithelial–mesenchymal transition (EMT
) by underregulating the STAT3 pathway
[8][86]. LncRNA uc.283-plus
[9][87], LncRNA CRNDE
[10][88], and LncRNA XIST
[11][89] are all known to be overregulated in glioma stem cells. Two lncRNAs, PRNCR1 and PCGEM1, are found to be overexpressed in different prostate cancers and cause the androgen receptor (AR)–associated transcriptional mechanisms to induce the growth of prostate cancer
[12][13][90,91]. Maternally expressed gene 3 lncRNA (MEG3) was observed to stimulate p53 and promote p53 signaling, as well as increasing p53 associations with the promoters of its target genes
[14][92]. Recurrent hypermethylation in the MEG3 promoter is commonly seen in human tumors, such as pituitary cancer
[15][93], meningioma
[16][94], and leukemia
[17][95]. Higher expression of MEG3 works in a tumor suppressive manner by oppressing cell proliferation in meningioma and HCC cell lines
[18][96].
2. The Interplay of LncRNAs in CSCs Signaling Pathways
2.1. NOTCH Signaling Pathway
NOTCH signaling is a majorly conserved signaling pathway that carries out juxtracrine signaling between cells. NOTCH signaling takes place by the binding of two jagged ligands (Jagged1; Jagged2) and a transmembrane delta-like ligand either 1, 2, 3, or 4 (DLL1, DLL2, DLL3, DLL4) and one cell to a NOTCH receptor either 1, 2, 3, or 4 (NOTCH1, NOTCH2, NOTCH3, NOTCH4) on the neighboring cell. In the endoplasmic reticulum, NOTCH is synthesized and glycosylated by the soluble ER enzymes. In the Golgi, the S1 cleavage generates a heterodimer of NOTCH extracellular domain (NECD) and transmembrane NOTCH intracellular domain (TM-NICD). It then is transported to the plasma membrane to permit ligand binding. This binding is modulated by Deltex and obstructed by the activity of NUMB. After this, the S2 cleavage takes place, the NECD is cleaved from the TM-NICD domain by TNF-α ADAM metalloprotease converting enzyme (TACE) ADAM17 or a disintegrin and metalloproteinase domain-10 (ADAM10). In the signal-sending cell, the NECD stays confined to the ligand and experiences further recycling. Meanwhile, in the signal-sending cell, S3 cleavage is carried out by γ-Secretase, which releases the NICD segment. It is then translocated to the nucleus, where it associates with transcriptional regulators to instigate transcription of the NOTCH target genes.
NOTCH signaling plays a prominent role in the modulation of cell-fate specification, and progenitor cell differentiation, maintenance, and proliferation during organogenesis as well as during the early development of the hematopoietic cells and Central Nervous System
[19][20][97,98]. In stem cell niches, it is crucial in the differential activities of some stem cells, stem cell proliferation, and their interaction with the microenvironment. The carcinogenicity of the pathway was first observed in human T-cell acute lymphoblastic leukemia (T-ALL), where, due to the release of N1ICD, NOTCH-1 was seen to be activated
[21][99]. Since then, abnormal activation of the NOTCH family led to tumorigenesis of various cancers, such as pancreatic
[22][100], breast
[23][101], and cervical cancer
[24][102]. This pathway is observed to operate as both oncogenic and tumor-suppressive, this is dependent on the tissue type, cancer cell type, as well as the stage involved. NOTCH-1 stimulates cancer development at an early stage of cervical cancer, while it represses cancer growth at a later stage. As an oncogene, NOTCH is seen to be overexpressed in T-ALL, pancreatic, breast, gastric, and colon cancer
[22][23][25][26][27][100,101,103,104,105]. As an example, mutations leading to activated NOTCH1 can stimulate the development of T-ALL
[28][106]. PCSCs were observed to have increased levels of NOTCH1, NOTCH3, Jag1, Jag2, as well as the NOTCH target gene Hes1; this was responsible for the maintenance of CSCs stemness
[29][107]. Meanwhile, its expression is downregulated in myeloid malignancies, skin, non-small cell lung cancer, liver, prostate, and certain breast cancers
[30][31][32][33][34][108,109,110,111,112] where it plays a tumor-suppressive role. It was observed that NOTCH-1 activation is responsible for inducing the MMP-2 and MMP-9 expression and stimulation while lowering the regulation of NOTCH-1 involved in lowering the MMP-2 and MMP-9 activation to hinder cell progression in breast cancer and pancreatic cancer cells
[35][113].
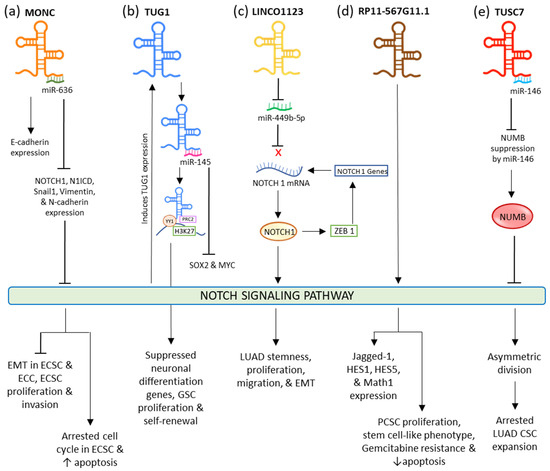
In endometrial carcinoma (EC), tumor-suppressive MONC sponges oncogenic miR-636 to negatively regulate it. The upregulation of this lncRNA restricted the expression of NOTCH1, N1ICD, Vimentin, Snail1, and N-Cadherin, while the expression of E-Cadherin was fostered. This eventually inhibited the NOTCH signaling pathway and EMT resulting in suppressed proliferation and invasion, and induced apoptosis, and aided the arrest of the cell cycle at the G0/G1 phase in endometrial cancer cells and cancer stem cells (ECCs and ECSCs) (
Figure 12a)
[36][114]. Increased expression of lncRNA TUG1 (Taurine Upregulated Gene 1) is seen in different types of cancers such as bladder carcinoma, stomach cancer, and bone sarcoma
[37][38][39][115,116,117], while its decreased expression is seen in non-small cell lung cancer (NSCLC)
[40][118]. This lncRNA is credible for sustaining the stemness properties and proliferation of glioma stem cells (GSC) in glioblastoma via neutralizing miR-145 by sponging it in the cytoplasm and modulating transcription factors MYC and SOX2. In the nucleus, it forms a polycomb structure with YY1, PRC2, and H3K27, from which histone H3K27 undergoes locus-specific methylation via the activity of YY1 to limit the neuronal differentiation genes (
Figure 12b). In lung adenocarcinoma (LUAD), ZEB1, an EMT transcriptional factor, activates LINC01123 and NOTCH1. LINC01123 functions by sponging miR-449b-5p and blocking its inhibitory effect on NOTCH1 mRNA; this results in the expression of NOTCH1, thus mediating the promotion of the NOTCH1-dependent NOTCH signaling pathway. This results in the lncRNA facilitating increased stemness, proliferation, migration, malignant transformation, and EMT in LUAD (
Figure 2c)
[41][119]. In pancreatic cancer, RP11-567G11.1 is observed to upregulate the NOTCH signaling pathway and the expression of its downstream components. This aberrant activation resulted in the progression of the cell cycle, PCSCs proliferation, tumorigenesis, and decreased apoptosis. RP11-567G11.1 also mediated the chemoresistance to antimetabolite Gemcitabine (
Figure 2d)
[42][120]. LncRNA TUSC-7 is responsible for inactivated NOTCH signaling in lung adenocarcinoma (LUAD) stem cells. This leads to the disruption in the symmetric division, causing the appointment of asymmetric division and ultimately arresting the CSC expansion.
Figure 12.
Interaction of different lncRNAs and lincRNA with NOTCH signaling pathway.
It does so by inactivating the miR-146-mediated repression of NUMB, which plays a significant role in restraining the NOTCH signaling. It is responsible for restraining the ligand from binding by hindering the NOTCH receptor’s endosome transportation to the plasma membrane. When TUSC-7 is sponged to miR-146, its degradation toward NUMB is abolished, resulting in the inactivation of NOTCH signaling (
Figure 12e)
[43][121].
2.2. WNT Signaling Pathway
WNT signaling significantly participates in different biological activities, such as cell proliferation, differentiation, tissue regeneration, and organogenesis
[44][45][46][47][48][122,123,124,125,126]. This pathway is split into β-Catenin-dependent and β-Catenin-independent signaling; the former is known as the canonical pathway which is the WNT/β-Catenin signaling pathway and the latter is known as the non-canonical pathways comprised of WNT/calcium signaling and WNT/planar cell polarity [PCP]-signaling pathways
[49][50][127,128].
The canonical WNT signaling pathway is initiated when WNT-associated ligands link to Frizzled, a transmembrane receptor and the low-density lipoprotein receptor-related protein (LRP). Phosphorylation of LRP is then carried out, due to which Dishevelled proteins (DVL) become polymerized at the plasma membrane. This further hinders the destruction complex; comprising APC, AXIN, and GSK3β. This leads to the accumulation of β-Catenin in the cytoplasm, which then undergoes nuclear translocation. Once inside, β-Catenin is associated with the T-cell factor (TCF) and lymphoid enhancer factor (LEF); it functions by regulating multiple cellular mechanisms, thus behaving as a transcriptional switch
[51][129].
In the non-canonical calcium-signaling pathway, WNT binds to DVL to stimulate phospholipase C (PLC) which releases calcium ions. These intracellular ions stimulate the downstream protein kinase C (PKC) and calcium/calmodulin-dependent protein kinase II (CaMKII) which in turn positively affect the nuclear factor (NFAT)
[52][130]. In the non-canonical PCP-signaling pathway, the planar cell polarity is triggered by WNT–Frizzled receptor associations that activate DVL. Activated DVL then modulates RHOA, RAC, and CDC42 GTPases. It further triggers the JUN N-terminal kinase, which then activates nuclear factor-dependent transcription of activator protein-1 and stimulated T cells after nuclear transfer.
The canonical pathway is mostly known as regulating cell proliferation, while noncanonical pathways manage cells’ polarity and movement. However, both signaling pathways participate in tumorigenesis and can also become involved in different cellular processes
[53][131]. In the same way, adenomatous polyposis coli (APC) and β-Catenin not only regulate cell proliferation but are also involved in cell-to-cell adhesion
[54][132]. In colon cancer, R-SPONDIN/LGR5/RNF43 mutations are linked with the deregulation of WNT leading to tumor development
[51][129]. Approximately 92% of sporadic colorectal cancers are observed to have at least an alteration in the regulators of the WNT pathway
[55][133]. In a wide range of breast cancers, plummeted levels of nuclear β-Catenin are reported, as well as seeing regular plummeted expression of the receptors and ligands. However, unlike colorectal cancer, the development of breast cancer is not due to the genetic regulation of the signaling components of the WNT pathway, but is due to other factors such as epigenetics
[56][134]. In different types of leukemias, the deregulated WNT pathway leads to increased WNT activity, eventually triggering leukemogenesis. In liver-cancer stem cells (LCSC), activation of canonical WNT signaling is linked with elevated self-renewal capacity of cells
[57][135]. WNT signaling was seen to elevate the expression of a colon cancer SC marker CD44v6, which resulted in an aggravated metastatic capacity of the CSCs
[58][136]. In the breast cancer niche, the cross talk of Periostin with WNT1 and WNT3a stimulates WNT signaling, resulting in the sustained phenotype of cancer stem cells
[59][137].
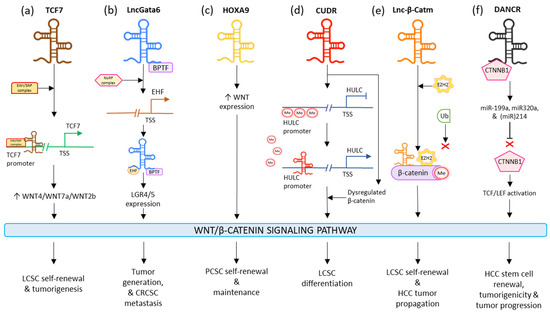
LncTCF7 appoints the SWI/SNF to the TCF7 promoter and improves its expression to surge the levels of WNT7a/WNT4/WNT2b, which activates the WNT signaling pathway. The lncTCF7-associated WNT stimulation results in the maintenance of the self-renewal and tumorigenic ability of LCSCs (
Figure 23a)
[57][135]. LncGata6 enhances tumor generation and metastasis in colorectal CSCs. LncGata6 associates with Bromodomain PHD Finger Transcription Factor (BPTF) and adopts the Nucleosome Remodeling Factor (NURF) complex to trigger ETS homologous factor (EHF) transcription, which makes up the lncGata6–BPTF–EHF axis to generate LGR4/5 expression and eventually activate the WNT signaling pathway (
Figure 23b)
[60][138]. LncRNA HOTTIP is observed to drive the stimulation of the WNT/β-Catenin signaling pathway; this induces the cell generation and chemoresistance in osteosarcoma. It is also responsible for elevating the stemness of PCSCs by regulating the WNT pathway. HOXA9 regulates the stimulation of the WNT pathway in PCSCs by promoting WNT. HOXA9 targeted stem cell transcription factors (SOX2, Nanog, OCT4, and LIN28) and markers (ALDH1, CD44, and CD133) in order to sustain PCSC properties such as inducing CSC expansion and self-renewal (
Figure 23c)
[61][139]. Interestingly, LncRNA CUDR stimulates lncRNA HULC by obstructing the methylation of the HULC promoter; additionally, it dysregulates β-Catenin thus promoting malignant differentiation in human-liver stem cells (
Figure 23d)
[62][140]. LincRNA-p21 is seen to reduce the activity of β-Catenin signaling, ultimately mediating the negative regulation of self-renewal, viability, and glycolysis of colorectal-cancer stem cells (CRCSCs)
[63][141]. The levels of lncRNA H19 are observed to plummet in bladder cancer tissues. H19 is complexed with the enhancer of zeste homolog 2 (EZH2), such that this association results in the activation of WNT/β-Catenin. This activation reduces the expression of E-cad and increases tumor metastasis. Upregulated H19 is also responsible for promoting bladder-cancer cell migration
[64][142]. Lnc-β-Catm catalyzes the methylation process of β-Catenin, by binding to it along with methyltransferase EZH2. This limits the ubiquitination of β-Catenin and thereby stabilizes it, which eventually drives the activation of the WNT/β-Catenin signaling pathway. This activation promotes self-renewal and maintenance of LCSCs and tumor progression of HCC (
Figure 23e)
[65][143]. In HCC, microRNAs miR214, miR320a, and miR-199a are seen to have a suppressive effect on CTNNB1; DANCR blocks this suppression by associating with tethering to CTNNB1. This induces the activation of TCF/LEF by β-Catenin, which guides the positive regulation of HCC stem cells’ self-renewal, tumorigenicity, and cancer progression (
Figure 23f)
[66][144]. In gastric cancer, HOTAIR was seen to be majorly regulated in cisplatin-resistant gastric cancer cells and tissues. It was observed that the higher expression of HOTAIR promoted gastric-cancer cell proliferation, enhanced the transition of cell cycle G1/S, as well as decreased cancer cell apoptosis by activating WNT/β-Catenin signaling. Moreover, the lncRNA was also responsible for binding and inhibiting miR-126 expression; this resulted in the promotion of VEGFA and PIK3R2 expression and activation of the PI3K/AKT/MRP1 pathway
[67][145].
Figure 23.
Influence of different lncRNAs on WNT/β-Catenin CSC signaling pathway.
2.3. STAT Signaling Pathway
In humans, the STAT family incorporates STAT-1, 2, 3, 4, 5A, 5B, and 6
[68][146]. Among these seven proteins, STAT-3 and STAT-5 demonstrate the highest association with tumor progression. Their activation regulates a broad range of functions, such as cell cycle progression, proliferation, apoptosis, angiogenesis, as well as immune evasion
[69][70][71][147,148,149]. STAT-3 is primarily responsible for tumor growth, proliferation, and sustenance due to its activity in stromal cells, immune cells, and the tumor microenvironment
[72][73][74][75][76][77][150,151,152,153,154,155]. The negative modulation of STAT-3 activation is carried out by protein repressors of stimulated STATs (PIAS), tyrosine phosphatases, and oppressors of cytokine signaling (SOCS)
[78][156].
Abnormal activation of the pathway is seen in CSCs from different cancers such as breast, blood, glia, and prostate. In prostate cancer, stem-like cells were seen to overexpress various genes linked to JAK/STAT signaling pathway, such as IFNK, IFNGR, IL6, CSF2, and STAT1
[79][157], and stimulate STAT3 to prominently regulate JAK/STAT signaling molecules in mammalian CSC-like cells
[80][158]. In glioblastoma, obstruction of STAT3 in CSCs shuts down proliferation, as well as lowering the expression of OLIG2 and NESTIN (neural stem cell gene), and enhancing the expression of βIII-Tubulin, which acts as a neuronal differentiator
[81][159]. Thus, in glioblastoma, JAK/STAT signaling is significant for CSC proliferation and is responsible for positively regulating glioblastoma stemness. In acute myeloid leukemia (AML), activated JAK/STAT signaling led to the growth and sustenance of CSCs
[82][160].
Abnormal STAT3 activation, because of tyrosine 705 phosphorylation signaling, led to the oncogenesis and induction of malignancy
[83][84][85][86][161,162,163,164]. Activation of STAT3 in the tumor causes transfer of indicators from the different growth factors and cytokines
[87][165] which in turn induces certain target genes such as Cyclin-D, c-Myc, and CDC25A. These factors ultimately activate the proliferation of cells, suppress the apoptotic genes
[88][166], and upregulate anti-apoptotic genes such as Beta2-Macroglobulin, B-Cell CLL/Lymphoma-2 (BCL2), and BCLXL
[89][167]. The induction of STAT3 by IL-6
[90][168] or stress factors
[91][169] leads to progress in the self-renewal of prostate CSCs and tumor-propagation
[92][170]. VEGF binds to VEGFR-2/JAK2/STAT3, leading to the activation of STAT3 and upregulation of Myc and SOX2. This eventually results in enhanced self-renewal of breast and lung CSC. Thus, apart from angiogenesis, VEGF is also responsible for CSCs’ self-renewal and tumor-initiating via the VEGFR-2/STAT3 signaling
[93][171].
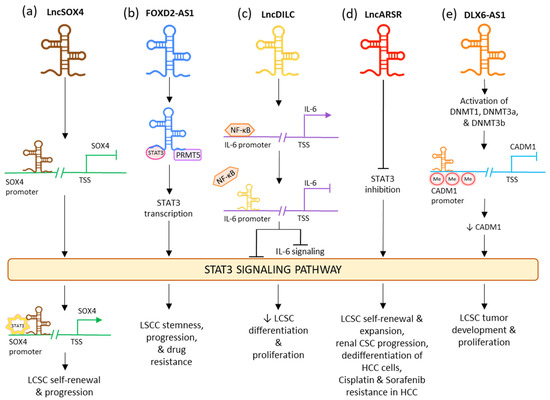
LncRNA downregulated in liver-cancer stem cell (lnc-DILC) acts as a tumor suppressor by declining the proliferation rates of CSC and differentiation of the LCSCs
[94][172]. It also has an intense hindering effect on the autocrine pathway of IL-6/JAK2/STAT3 signaling
[95][173]. This lncRNA lowers the expression of STAT3 as well as IL-6 and also limits the translocation of STAT3 to the nucleus. It also acts by inhibiting the NF-κB-mediated induction of IL-6. In various tumor stem cells, IL-6 is highly expressed by the NF-κB signaling pathway which is triggered by growth factors such as TNF-α and IL-1β (
Figure 34c)
[96][97][174,175]. LncRNA DILC and lncSOX4 are well known for controlling CSC properties by the STAT3 pathway in liver cancer. Lnc-DILC can also regulate the JAK2/STAT3 signaling. Overexpression of lnc-DILC lowers the elevated levels of phospho-STAT3 protein, reduces the translocation of STAT3 to the nucleus, and oppresses the transcriptional activity of STAT3-responsive segments. In CSC spheroids, the decreased regulation of lnc-DILC activates TNF-α and IL-1β-triggered IL-6 expression. Thus, this lncRNA can synchronize the crosstalk between the inflammatory signaling and the autocrine IL-6/STAT3 pathway to affect the expansion of LCSC
[98][176]. On the other hand, lncSOX4 increased regulation has been associated with critical liver cancer. It is positively regulated in the CD133+ liver cancer cell population and CSC spheroids and is crucial for the self-renewing activities and tumorigenic capability of the liver CSCs. LncSOX4 crosstalks with STAT3 and inducts it to the promoter region of SOX4, stimulating the H3K4me3 and H3K27ac alterations to increase the SOX4 promoter stimulation and thus SOX4 expression. The lncSOX4/STAT3-dependent SOX4 expression is significant for sustaining liver CSC proliferation (
Figure 34a)
[99][177]. Upregulated FOXD2-AS1 was identified in maintaining the stemness in laryngeal squamous cell carcinoma (LSCC) and decreasing the reactions to chemotherapy drugs, while its under-regulation had opposing consequences. FOXD2-AS1 is complexed as a scaffold with STAT3 and PRMT5, stimulating the transcription STAT3, which is important for maintaining tumor stemness and enabling chemotherapeutic resistance. Thus, FOXD2-AS1 acts as an upstream activator of STAT3 and aids cancer progression (
Figure 34b)
[100][178]. LncARSR overexpression is seen in CD24+/CD133+ LCSCs as well as in hepatoma spheres augmented with CSC. Its upregulation causes blockage of STAT3 inhibition, thus activating the STAT3 signaling. STAT3 also plays a downstream role in regulation by lncARSR in HCC cells. LncARSR is responsible for LCSC expansion by rising the dedifferentiation of hepatoma cells and increasing the self-renewal, progression, and tumorigenesis capacity of LCSCs. It also aids drug resistance and tumor recurrence (
Figure 34d)
[101][179]. In early LSCC, the levels of lncRNA PTCSC3 in plasma are low. The increased expression of PTCSC3 causes repression of LSCC cell proliferation; thus, the lncRNA acts as a tumor suppressor. In UM-SCC-17A cells, PTCSC3 hindered STAT3 and HOTAIR’s expression, while STAT3 was responsible for increasing the expression of HOTAIR. Thus, it can be stipulated that the oppressive effect of PTCSC3 on tumor progression can be connected to lncRNA HOTAIR
[100][178]. The downregulation of LncRNA DLX6-AS1 may cause an inhibitory effect on the methylation of the CADM1 promoter. This may lead to the inactivation of the STAT3 signaling pathway. Upregulated CADM1 results in the suppression of the stem cell characteristics of liver CSCs
[102][180]. In HCC, overactivated lncRNA DLX6-AS1 confers the methylation of CADM1 promoter via activation of methyltransferases DNMTs and expression of SOX4 in LCSCs and thus upregulating the STAT3 signaling pathway. This led to decreased expressions of SOX2, OCT-4, CD13, and CD133 in LCSCs and induced tumor formation and proliferation (
Figure 34e)
[102][180].
Figure 34.
Influence of different lncRNAs on STAT3 CSC signaling pathway.
2.4. Hedgehog Signaling Pathway
Abnormal induction of Hedgehog is linked with the proliferation of cancer cells, neoplastic transformations, malignancy, metastasis, and multi-drug resistance of a wide range of tumors. When Hedgehog is not present, PTCH1 localizes in the cilia and avoids the membrane localization and stimulation of SMO. SMO after internalization undergoes degradation. The full-length GLI (GLIFL) is altered by protein kinase A (PKA), glycogen synthase kinase-3 (GSK3), casein kinase 1 (CK1), and the E3 ubiquitin ligase β-TrCP. It then undergoes cleavage to form transcriptional oppressor GLIR. The stimulated form of GLI (GLIA) is repressed by SUFU. Then, GLIR undergoes nuclear translocation and hinders the expression of its target genes. When the Hedgehog binds to PTCH1, it internalizes and carries out the translocation and induction of SMO. GLIFL avoids phosphorylation by PKA, GSK3, and CK1, which causes activation of GLI (GLIA). GLIA then undergoes nuclear translocation where it activates the expression of Hedgehog target genes
[103][181].
The activated Hedgehog pathway is involved in the growth, survival, migration, and proliferation
[104][182] of a wide range of cancers such as multiple myeloma, pancreatic adenocarcinoma
[105][183], breast cancer
[106][184], and chronic myelogenous leukemia (CML)
[107][185]. The first evidence of deregulated Hedgehog signaling and cancer was seen in Gorlin syndrome, which is caused due to the inducing mutation in the Patched gene
[108][186]. Patients with this syndrome display different cancer types such as basal cell carcinomas (BCCs)
[109][187], medulloblastoma
[110][111][188,189], and rhabdomyosarcoma
[112][113][190,191]. The paracrine stimulation of the Hedgehog pathway is seen in different tumor types such as prostate
[114][192], pancreatic
[115][193], colorectal, and esophageal cancers
[116][194]. In chronic myelogenous leukemia (CML), the loss of SMO impaired the self-renewal capacity of the hematopoietic stem cells and hindered the initiation of CML by the oncoprotein: BCR-ABL1
[117][195]. Sonic Hedgehog, SMO, and GLI1 are highly aggravated in CML suggesting that activation of the Hedgehog pathway is a significant activator of CML progression
[118][196]. Hedgehog inactivation in MCF-7-derived CSCs caused a reduction in the cells via the downregulation of OCT4, NESTIN, and Nanog, suggesting that Hedgehog signaling in breast CSCs upregulates stem cell markers to retain a self-renewing phenotype
[119][197].
This signaling plays a crucial role in the sustenance of colon CD133+ CSCs, which show the increased gene expression levels of GLI1, PTCH1, GLI2, SHH, and HHIP concerning all CD133- cells in colon carcinoma
[120][198].
Yes-associated protein 1 (YAP) is responsible for upregulating the transcription of lncRNA BCAR4, which interacts with SNIP1 and PNUTS to regulate the p300-dependent histone acetylation carried by H3K27ac of the non-canonical Hedgehog and GLI2 transcriptional cascade. This stimulates the transcription of enzymes HK2 and PFKFB3; thus, allowing BCAR4 to promote metastasis and invasion in triple-negative breast cancer as well as reprogram glucose metabolism in favor of the CSCs (
Figure 45a)
[121][199]. LncRNA-cCSC1 is seen to be overexpressed in colorectal cancer cells (CRC) and CRCSCs. The exhaustion of lncRNA-cCSC1 prominently hindered the self-renewal activity of the CRCSCs and decreased their chemo-drug resistance to 5-fluorouracil. Meanwhile increased expression of lncRNA-cCSC1 led to triggered self-renewal of CSCs as well as assisting in 5-fluorouracil drug resistance. Abnormal expression of this lncRNA resulted in the transfiguration of SMO and GLI family zinc finger 1 (GLI1) expression in the Hedgehog signaling pathway (
Figure 45b)
[122][200]. LncRNA lncHDAC2 is overexpressed in LCSCs. In the LCSC niche, it is associated with histone deacetylase-2 (HDAC2). LncHDAC2 regulated PTCH1 downregulation to stimulate Hedgehog signaling, thus increasing the self-renewal activity of LCSCs (
Figure 45c)
[123][201]. Overexpression of ASAP1-IT1 is seen in cholangiocarcinoma (CCA) tissues and cells. This lncRNA is responsible for activating the Hedgehog pathway via the increased regulation of SMO and GLI1, thus inducing cell proliferation, cell migration, and the progression of EMT
[124][202].
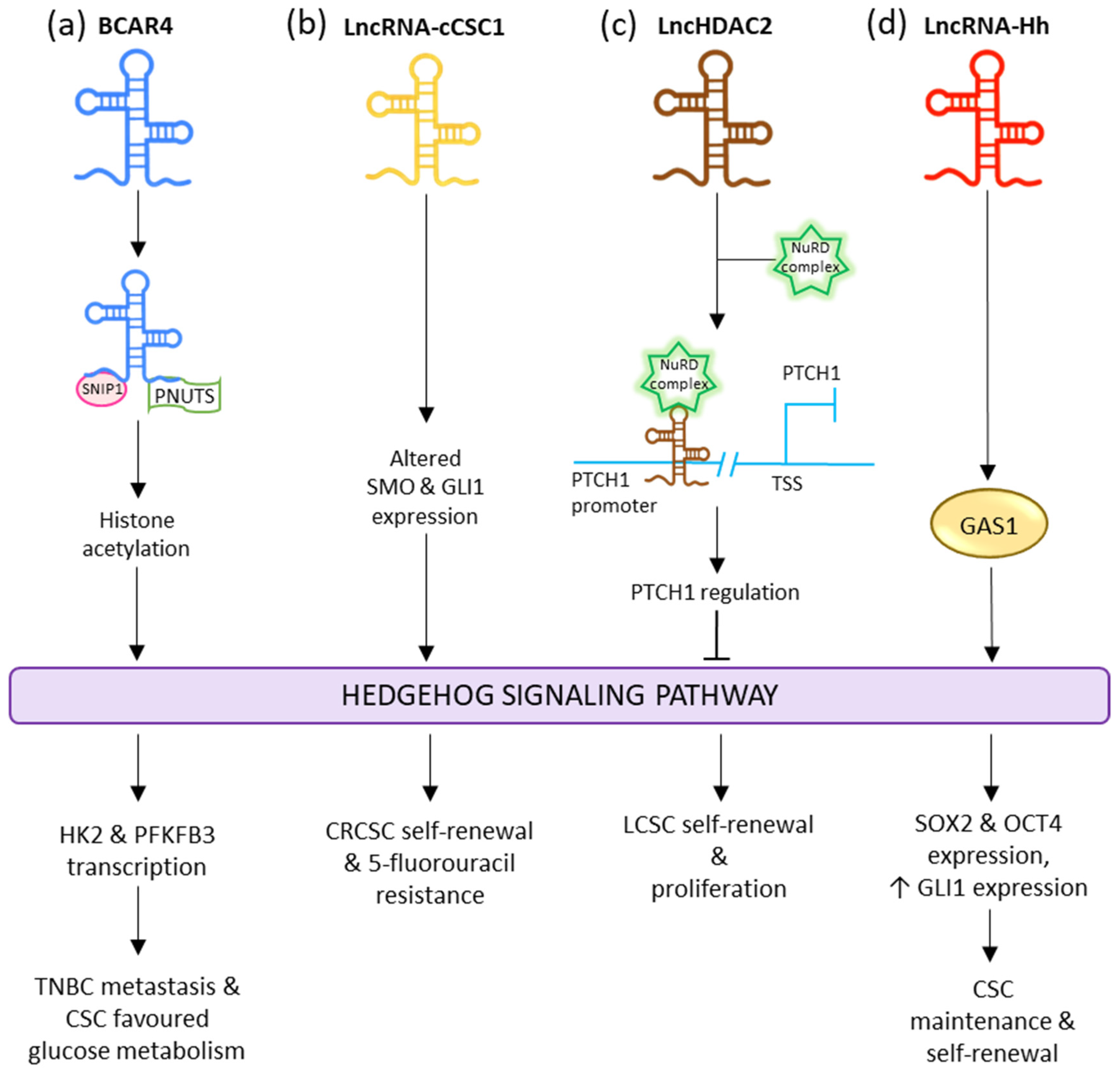
Figure 45.
Influence of different lncRNAs on Hedgehog CSC signaling pathway.
LncRNA EGOT acts as a significant modulator in breast cancer. In gastric cancer, with elevated expression, it acts as an oncogene. EGOT-regulated Cyclin D1 expression in GC is modulated by the Hedgehog pathway. Its upregulation is related to gastric cancer cell growth, cell cycle progression, and lymphatic metastasis
[125][203]. LncRNA HCG18 participates as an oncogenic lncRNA in nasopharyngeal cancer (NPC) progression. It does so by modulating WNT/β-Catenin signaling and the Hedgehog pathway. Upregulation of HCG18 is associated with tumor progressive effects via the stimulation of cell proliferation and metastasis
[126][204]. LncRNA-Hh is known to target GAS1 to induce the activation of Hedgehog signaling. The activated Hedgehog signaling raises the expression of GLI1 and stimulates the expression of SOX2 and OCT4 to regulate CSC maintenance and self-renewal (
Figure 45d)
[127][205].