1. Introduction
The β superfamily of transforming growth factors is a numerous group of evolutionarily conserved ligands involved in the regulation of cellular, physiological, and pathological processes. Signalling by TGFβ influences embryonic development and tissue homeostasis, including angiogenesis, tissue regeneration, modulation of the immune response, extracellular matrix remodelling, cell mobility, and apoptosis in physiological and pathological conditions, especially during development, tumour progression, and metastasis
[1][2][3][4][5][1,2,3,4,5].
The pleiotropic activity of the signalling pathways induced by TGFβ superfamily factors reflects the complexity of the signal transmitted. This complexity is observed in TGFβ ligands, more than 30 of which have been identified so far. TGFβ occurs in human cells in three isoforms: TGFβ1, TGFβ2, and TGFβ3. Their amino acid sequence similarity is 71–80%
[6]. TGFβ isoforms are synthesised as a pre-protein. The native form is dimeric, and the primary structure of active ligands contains a motif of 6 to 12 cysteine residues, referred to as the cystine knot (CK). The presence of the cystine knot is responsible for the formation of homo- and heterodimers of TGFβ factors, and their active form has a molecular weight of approximately 25 kDa.
TGFβ1 is synthesised as a 390 amino acid proprotein, which undergoes post-translational processing. Amino acids 1–29 form the signal peptide, amino acids 30–278 form the latency-associated peptide LAP, whereas the remaining amino acids 279–390 form TGFβ1. The LAP and TGFβ1 chains remain noncovalently linked during storage in the extracellular matrix, which keeps TGFβ1 inactive. The sequence of the protein is available in the EBI database (P01137), and the structure (1KLA in the Protein Data Bank in Europe) of the homodimer TGFβ1 (
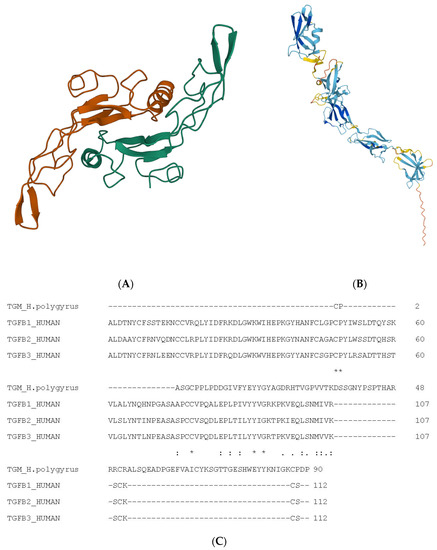
Figure 1A) was determined by Hinck et al. 1996
[7].
Figure 1. Structure of TGFβ1 and nematode-derived mimic TGFM. (
A) Structure of TGFβ1. (
A) Used Mol* to create visualisation
[8]. Alpha helices are indicated as coils, whereas beta-pleated strands are indicated as arrows. (
B) Structure of TGFM. (
B) Also used Mol* for visualisation. Alpha helices are indicated as coils, whereas beta-pleated strands are indicated as arrows. (
C) Multiple sequence alignment of TGFβ isoforms and nematode-derived mimic TGFM. The ‘*’ indicates an identical amino acid while the ‘:’ indicates a similar amino acid.
Canonical cell signalling induced by ligands belonging to the TGFβ superfamily is mediated by highly specific serine and threonine kinase receptors, which are expressed by most types of human cells. TGFβ receptors are transmembrane glycoproteins with an N-terminal region responsible for ligand binding, a single transmembrane fragment, and a C-terminal cytoplasmic region in which the kinase domain is located. Activation of the canonical TGFβ pathway occurs when the dimeric TGFβ ligand binds to the TGFβ type II receptor dimer, which, having constitutive kinase activity, undergoes autophosphorylation. The TGFβR1 receptor dimer is bound to the TGFβ–TGFβR2 complex, and serine and threonine residues in the repeating glycine and serine residues (GS region) are then phosphorylated by the activated TGFβR2 receptor. The signal is then transmitted through the TGFβ Ina receptors of the Smad cytoplasmic proteins. Canonical signalling of TGFβ is negatively regulated by the Inhibitory Smads (I-Smad) proteins Smad6 and Smad7. The I-Smad proteins, unlike the R-Smad and Co-Smad proteins, do not contain the MH1 domain responsible for DNA sequence recognition and transcriptional activity. The MH2 domain determines the binding of the Smad7 protein to TGFβ receptors, thus competing with the R-Smad proteins. Similarly, TGFβ cell signalling is inhibited by the Smad6 protein, which, by binding to the Smad4 protein, reduces the formation of Smad1–Smad4 complexes
[9][10][11][12][9,10,11,12].
Many Faces of TGFβ
Due to the influence of TGFβ on an array of diverse cellular functions including cell growth, differentiation, adhesion, migration, and apoptosis, perturbations of the TGFβ signalling pathways are involved in the progression of various tumours. TGFβ is a multifunctional cytokine that acts in a cell- and context-dependent manner as a tumour promoter or tumour suppressor. This phenomenon is known as the TGFβ paradox
[13]. In healthy cells and early-stage cancer cells, TGFβ ligands stimulate signalling pathways leading to the expression of genes involved in tumour suppression, inhibition of proliferation, stimulation of differentiation, induction of apoptosis or autophagy, elimination of inflammation, and suppression of angiogenesis. In contrast, advanced tumours produce excessive amounts of TGFβ, which contributes to tumour growth, invasion and metastatic spread, and drug resistance
[5].
The pleiotropic function of TGFβ in tumour development is due to the interaction of this cytokine with various signalling pathways. Activation of the TGFβ type II receptor (TβRII) and, in turn, type I receptor (TβRI) by TGFβ1 transduces signals through receptor-regulated Smads (Smad2/3) and common-partner Smad (Smad4), leading to the transcriptional regulation of target genes. Reflecting its diverse and complex functions in cancer cells, TGFβ upregulates some autophagy-related genes in a Smad4-dependent fashion. Thus, certain hepatocellular carcinoma cell lines undergo cell cycle arrest and apoptosis in response to TGFβ
[14].
However, a number of noncanonical TGFβ signalling pathways are responsible for unexpected signalling outcomes or even opposing biological outcomes of TGFβ signalling in the same cells
[15]. The activated TGFβ complex transfers signals through molecules such as receptor-associated factor 4 (TRAF4), TRAF6, TGFβ-activated kinase 1 (TAK1), p38 mitogen-activated protein kinase (p38 MAPK), p42/p44 MAPK, phosphoinositide 3-kinase Pi3K/AKT, extracellular signal-regulated kinase (ERK), Rho-like GTPase signalling pathways, JUN N-terminal kinase (JNK), or NF-κB to reinforce or decrease downstream cellular responses
[16]. Some of these pathways could be transducers of the TGFβ signal to Smads
[17].
Furthermore, the regulation of TGFβ signalling in cells can include the intracellular distribution of the receptors, composition of the receptors, and expression of accessory molecules. For example, mature alternatively activated macrophages (M2) induced by IL-4 require the presence of glucocorticoids (GCs) to express TGFRII on the cell surface to become permissive to the TGFβ
[18].
TGFβ is involved in interactions between cancer cells and the host immune system termed as “immunoediting” and summarised in the three “E’s” theory: elimination, equilibrium, and escape. Because of (i) genetic instability and tumour heterogeneity and (ii) immune selection pressure, tumour cells become progressively capable of avoiding immune destruction during carcinogenesis
[19][20][21][19,20,21]. Evading immune eradication is a prerequisite for neoplastic progression. The immune escape strategies of cancer may be classified into two main mechanisms. First, cancer cells may become invisible to the immune system. This can be achieved by losing or downregulating MHC and/or molecules involved in antigen presentation, thereby preventing their recognition by the immune system. Second, cells may “defend” themselves to become resistant to immune eradication. This can be achieved in several ways: by becoming resistant to apoptosis, expressing inhibitory ligands that deactivate immune cells, and/or inducing an immunosuppressive microenvironment, the TME
[19].
In cancer, TGFβ is recognised as one of the most important regulators in the tumour microenvironment (TME), which is a highly heterogeneous milieu consisting of different cell types. The TME includes the presence of immunosuppressive cell populations such as tumour-infiltrating myeloid cells, including myeloid-derived suppressor cells (MDSCs) and M2-like tumour-associated macrophages (TAMs). Likewise, the presence of immune regulatory populations, including regulatory T cells (Tregs), regulatory B cells (Bregs), and regulatory dendritic cells (DCregs), can mediate immunosuppression
[22]. These cell types beside cancer cells are not just sources of TGFβs; they seem to exploit autocrine or paracrine TGFβ for their expansion, polarisation, and behaviour towards a tumour-promoting role rather than tumour elimination
[22]. Tumour immune escape is complex, and a crucial aspect is promoting the expansion and activation of immature DCs. The immature DCs that uptake apoptotic and necrotic DCs convert into tolerogenic DCs (tDCs) with enhanced TGFβ secretion
[23]. tDCs display low expression of costimulatory molecules such as CD80 and CD86 as immature DCs; but they simultaneously have high expression of inhibitory molecules (TRAIL, PD-L1, DC-SIGN, and CTLA-4) and immunosuppressive molecules (e.g., TGFβ, IL-10, IL-27, NO, and IDO) and, in turn, support the Treg differentiation of type 1 Tregs (Tr1) in response to IDO, IL-10, and TGFβ.
Naïve CD4
+ T cells differentiate into CD25
+ Foxp3
+ Tregs (Tregs) in the thymus and into CD25
− Tregs including IL-10
+ Tr1, TGFβ
+ Th3, and Foxp3
+ cells (iTregs) in the periphery. Treg cells promote tumour growth and progression through multiple inhibitory pathways, and TGFβ is secreted at high levels by Treg cells in the tumour microenvironment. Blockade of TGFβ expressed on the surface of Treg cells using neutralising antibodies improved immunity to melanoma and suppressed the metastasis of pancreatic tumours in mice
[24][25][24,25]. Furthermore, TGFβ mediates T-cell suppression via programmed death-1 (PD-1) coinhibitory receptor 1 (immune checkpoint) upregulation through Smad3-dependent and Smad2-independent transcriptional activation in T cells and in the TME
[26].
In response to the TGFβ released by different cell types, including tumour cells in the TME, circulating monocytes that express high numbers of TGF- receptors migrate into the TME, where they differentiate into tumour-associated macrophages (TAMs). On these cells, TGFβ induces markers specific for M2-type macrophages with tumour supportive function
[13].
Other immune cells such as cytotoxic T cells (CTLs), natural killer (NK), and neutrophils involved in the anticancer response are also regulated by TGFβ. In the mouse system, TGFβ prevents the activity of cytotoxic CD8
+ T cells by inhibition of perforin, granzymes (GzmA, GzmB), interferon-γ (IFN-γ), and FasL expression
[27]. Correct NK signal transduction by NKG2D surface receptor–ligand binding culminates in the degranulation of NK cells to eliminate tumour cells. NKG2D ligands on tumour cells are downregulated, among other molecules, by TGFβ to escape NK-cell-mediated immune surveillance
[28]. TGFβ can also inhibit NK cell activity by decreasing IFN-γ production by these cells
[29].
Neutrophils are the main group of cells that infiltrate tumours, and they have a critical function in the immunosuppression of the TME as the tumour evolves. Similarly to M2, N2 neutrophils (N2 TANs) display protumourigenic activity, and the TGFβ within the tumour microenvironment induces N2 cells. TGFβ-stimulated N2 secretes different molecules that shape the TME. Neutrophils can be activated to display a stronger antitumour phenotype by blocking TGFβ
[30][31][30,31].
2. TGFβ Signalling in Helminths
Even such primitive animals as sponges and
Trichoplax produce molecules belonging to the TGFβ family
[32][39]. However, TGFβ seems to be unique to the animal kingdom. There are no documented members of the protein family in other kingdoms of living organisms. Several species of nematodes, both free-living and parasitic, have TGFβ signalling pathway components.
The best-described TGFβ pathway in nematodes is TGFβ signalling in Caenorabditis elegans. The TGFβ pathway plays fundamental roles in the development of this free-living roundworm. Five ligands (Ce-DBL-1, Ce-DAF-7, CeUNC-129, Ce-TIG-2, and Ce-TIG-3) and three receptors (Ce-DAF-1, Ce-DAF-4, and Ce-SMA-6) have been identified, and two signal pathways have been described for C. elegans.
The first pathway controls dauer larvae. Dauer larvae develop in response to unfavourable environmental conditions. Ce-DAF-7 influences entry and exit from the dauer stage and is a ligand for Ce-DAF-1 (type I) and Ce-DAF-4 (type II) receptors. Ce-DAF-8 and Ce-DAF-14 are components involved in signal transmission, whereas Ce-DAF-3 antagonistically acts towards them
[33][34][40,41]. The dauer form in free-living roundworms is hypothesised to be equivalent to the infective L3 stage of parasitic nematodes
[35][36][37][42,43,44]. It appears to be critical in the evolution of parasitism.
The second TGFβ-related pathway, called Sma/Mab, is involved in body size control, regulation of gland cell morphology, development of mail tail, immune defence, mesoderm differentiation, and reproductive ageing
[34][38][39][41,45,46]. Ce-DBL-1 is a ligand for Ce-SMA-6 and Ce-DAF-4 receptors in this pathway. SMA-2, SMA-3, and SMA-4 have been identified as Smad components and are involved in the Sma/Mab signal pathway
[34][41].
DAF-7, DAF-1, and DAF-4 are strongly conserved throughout nematode phyla. However, the regulators of the TGFβ pathway in
C. elegans have also been found in Clades IV and V (DAF-8 and DAF-3) or in Clade V (DAF-14) only
[40][47]. Parasitic nematodes independently evolved at least 15 times from free-living Nematoda
[41][48]. However, the role of the TGF pathway in the evolutionary origin of parasitism is unclear. One hypothesis states that the appearance of TGFβ in the evolution of nematodes had a key role in dauer formation and the emergence of invasive larvae from this stage
[42][49]. An alternative hypothesis considers TGF signalling to be primarily in development from iL3 to adult
[40][47].
Two TGF-related genes have been found in
Brugia malayi. This nematode is transmitted by mosquitoes and causes lymphatic filariasis in humans. Larval stages are found in the blood, whereas adults settle in lymphatic vessels. A transforming growth factor homolog-1 (Bm-tgh-1) shows similarity to genes encoding proteins in the dpp/Dbl-1 family. Bm-tgh-1 is mostly expressed during parasite growth when nematodes are present in the host
[43][50]. A second molecule, Bm-TGH-2 displays similarity to DAF-2 from
C. elegans and to human TGFβ. The similarity between Bm-TGH-2 and human TGFβ is restricted to the C-terminal domain with 38% identify. The gene encoding Bm-TGH-2 is maximally expressed in the microfilarial stages. As the parasite is exposed to various immune factors in the host blood, Bm-TGH-2 may play a significant role in modulating the immune response. Tgh-2 is expressed during the adult stage, in male as well as female species of
B. malayi. Recombinant Bm-TGH-2 binds to the TGFβ receptor, and this binding can be partially inhibited by human TGFβ
[44][51].
Brugia pahangi has a gene encoding a TGFβ receptor homologue termed Bp-trk-1
[45][52]. Bp-trk-1 shows similarity to TGFβ type I receptors and SMA-6 from
C. elegans [46][53]. Furthermore, filariae may be rich in molecules with similar structures to TGFβ. Antibodies that recognise the latent form of human TGFβ react with
B. malayi as well as many filarial worms including
Onchocerca volvulus,
Onchocerca gibsoni,
Onchocerca ochengi,
Onchocerca armillata,
Onchocerca fasciata,
Onchocerca flexuosa,
Wuchereria bancrofti, and
Dirofilaria sp. These antibodies may detect molecules with structural similarity to TGFβ1
[47][54].
Furthermore, three proteins in the hookworm
Ancylostoma caninum have been identified as TGF family members.
A. caninum develops in dogs and cats. Humans can also be infected with
A. caninum as an accidental host.
A. caninum DBL-1 (Ac-DBL-1) is involved in parasite growth regulation, and the protein has a similar amino acid sequence to
C. elegans DBL-1. The highest expression of Ac-dbl-1 is observed in the adult male stage; hence, DBL-1 from
A. caninum probably performs the same function as in
C. elegans—mail tail growth regulation. Another one, with the highest similarity to
C. elegans DAF-7, is called Ac-DAF-7 and regulates arrested development. Tissue-arrested L3 and reactivated L3 stages of
A. caninum are characterised by the highest Ac-daf-7 expression
[48][55]. In addition, human TGFβ can reactivate the tissue-arrested form of
A. caninum but cannot induce the development of environmental L3. It is possible that L3 possesses receptors for TGF molecules of mammalian origin
[49][56].
Haemonchus contortus is an important parasite found in the abomasum (true stomach) of goats and sheep. Two receptors belonging to the TGFβ family have been identified for
H. contortus. One has domains characteristic of a TGFβ type I: receptor Hc-TGFBR1, and one is a TGFβ type II receptor: Hc-TGFBR2
[50][51][57,58]. Both of them play a role in developmental processes of
H. contortus. Hc-TGFBR1 gene expression is observed during all developmental stages of the parasite. Interestingly, Hc-TGFBR1 is structurally more similar to human TGFBR1 than to DAF-1 from
C. elegans, suggesting a more similar function to a molecule of human origin
[50][57]. Furthermore, Galunisertib, a TGF-type I receptor inhibitor utilised in clinical trials involving cancer patients
[52][59], can block the transition of free-living L3 into the parasitic L4 of
H. contortus [48][50][55,57]. Hc-tgbr2 is expressed in all stages of the parasite, with the greatest levels of expression found in infective L3 and adult male species. Silencing of the gene reduced the level of the L3 to L4 transition in vitro
[49][56]. In addition, two TGF signalling ligands for
H. contortus have been characterised.
H. contortus DAF-3 (Hc-DAF-3) shows similarity to DAF-3 from
C. elegans and is classified as Co-Smad protein. Hc-daf-3 is expressed at the highest level by L3 and adult female species
[53][60]. Hc-TGH-2 is the second ligand described for
H. contortus. The Hc-tgh-2 gene is expressed by all developmental stages, with iL3 expressing the most. Hc-TGH-2 seems to play an important role in the transition from the free-living to parasitic stage, as well as in digestion, absorption, and reproductive development, based on the localisation of the ligand in parasite body
[54][61].
Molecules with similar sequences to TGF ligands have also been found in
Heligmosomoides polygyrus,
Necator brasilienisis, and
Teladorsargia circumcincta, although their functions have not been studied. The TGH-2 subfamily showed similarity to Ce-DAF-7, TGH-2 from
B. malayi and mammalian TGFβ. Interestingly, tgh-2 expression varies in four different Trichostrongyloid nematode parasites. The maximum expression occurs in adults of
H. polygyrus and
T. circumcincta, but only in L3 in
H. contortus and
N. brasiliensis [55][62].
Nematode TGFβ Mimic
Compounds that have a different structure but function similarly to TGF ligands have been reported in parasitic nematodes.
Heligmosomoides polygyrus is among the best understood of all nematode infections, and immune modulation has been clearly demonstrated
[56][79].
H. polygyrus has a direct life cycle
[56][79]. Adults live in the small intestine where they breed; eggs are laid by adult female species and excreted in the faeces. They develop through two moults into infective third-stage larvae. Infective larvae are ingested in natural infections but usually orally gavaged in experimental infections. Within 24 h, larvae have penetrated into the submucosa where they develop over the next ten days and undergo two moults before emerging into the lumen of the small intestine. About two weeks after infection, adult female species produce eggs that can be seen in the faeces.
H. polygyrus produces at least five molecules that modulate immune responses
[57][80], and these influence the immune response at several levels. One of them is a TGFβ mimic (
H. polygyrus bakeri TGFβ mimic, Hp-TGM), which influences the production of regulatory T cells. A family of ten Hp-TGMs are members of the complement control protein (CCP) superfamily, which also contains HpARI (
H. polygyrus bakeri alarmin release inhibitor) and HpBARI (
H. polygyrus bakeri binds alarmin receptor and inhibits). Hp-TGM has high immunomodulatory potential
[58][81] and consists of five atypical domains. The Hp-TGM family has no homology with mammalian TGFβ or other members of the TGFβ family
[59][82]. However, Hp-TGM can bind to TGFβ receptors and possesses TβRI and TβRII receptor-binding sites. Moreover, HP-TGM competes with mammalian TGFβ for TGFβ receptors
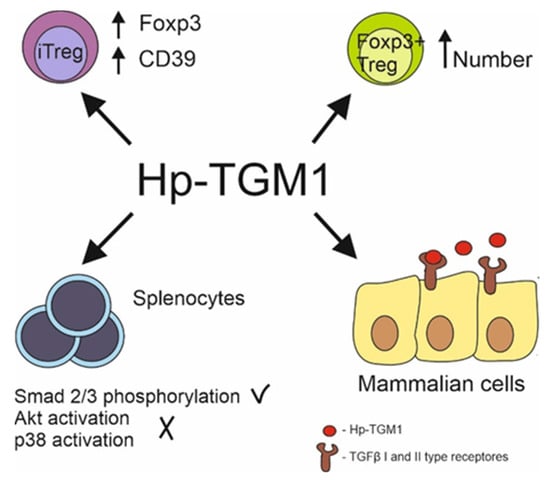
[60][83]. Although the TGM D3 domain and TGFβ bind the same residues in the TGFβ receptors, there is little sequence similarity between the molecules (
Figure 2).
Figure 2. Hp-TGM1 functions similarly to mammalian TGFβ despite lack of homologies between proteins. Hp-TGM1 binds to mammalian TGFβ I and II type receptors; induces Smad 2/3 phosphorylation with lack in Akt and p38 signal pathway activation in murine splenocytes; induces generation of mice and human Foxp3+ Treg cells; increases Foxp3 and CD39 expression in induced Treg same as mammalian TGFβ.