Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Maria Nikolova and Version 2 by Sirius Huang.
Liposomes are well-known nanoparticles with a non-toxic nature and the ability to incorporate both hydrophilic and hydrophobic drugs simultaneously. As modern drug delivery formulations are produced by emerging technologies, numerous advantages of liposomal drug delivery systems over conventional liposomes or free drug treatment of cancer have been reported.
- liposomes
- drug delivery
- cancer
- smart stimulus-responsive
- internal and external stimuli
1. Introduction
Cancer is thought to be a health problem with the leading cause of death [1]. The number of cancer cases is estimated to reach 21 million by 2030 [2]. Conventional chemo, radiotherapy, and hormone therapy are considered to be ineffective, due to low toxicity, non-specific distribution, and adverse effects [3]. Additionally, most conventional drugs suffer from poor pharmacokinetics, high toxicity, and reduced bioavailability. The field of nanotechnology has surged to a new height, which has inspired many researchers to produce a safer and more efficient drug delivery system using nanotechnology in the treatment of cancer therapy. The treatment of cancer with nanotechnology (nanooncology) has improved treatment efficacy by penetrating deep inside the body, where even the drug cannot reach [4]. The nanotechnologies hold numerous advantages in drug delivery systems, and a few of them include improving the in vivo pharmacokinetic process, enhancing the stability and longevity of the drug in blood circulation, and even modifying the carriers by targeting ligands on their surface for tissue or cell-specific delivery [5].
Significant achievements have occurred in the last few decades by applying injectable drug delivery systems (DDS) for cancer treatment. These advancements include the application of different nanoparticles, including liposomes, that conjugate various macromolecules. The nanocarriers, in the form of liposomes, polymeric nanoparticles, and even inorganic nanoparticles, can reach the interior of the cellular/molecular level and can detect the level of disease spread inside our body. Among the several nanoscale drug carriers, liposomes have demonstrated the greatest potential in various clinical applications [6]. Because of their similarities to biological membranes, liposomes offer excellent opportunities for the drug delivery of molecules into the target cells or subcellular compartments [7]. Lipid-based delivery systems offer cytoplasmic delivery by exploiting natural bio-functions, such as membrane fusion [8], because phospholipids are the main components of the biological cell membrane. Thanks to liposomes, it has become possible to increase the pharmacokinetics parameters of drugs, such as circulation time, controlled release, increased solubility, stability, and intercellular concentration [9].
Target-specific nanocarriers for drug delivery enhance the therapeutic efficacy of the loaded moiety by precisely targeting cancerous cells or tissues and preventing the drug from undergoing hepatic metabolism. To attain the desired pharmacotherapy, the nanovesicles should selectively release the drug at the targeted sites with minimum adverse effects. The cancer tissue targeting strategy can be both passive and active. Passive targeting mainly focuses on the pathological conditions of the disease, such as the difference in pore sizes among endothelial cells of cancer microvasculature that are larger than that of the structures of normal capillaries (an effect known as enhanced permeation and retention (EPR)). Only liposomes with diameters varying between 50 and 150 nm were able to avoid phagocytosis, enter blood vessels in the tumor microenvironment [10][11][10,11], and escape from capillaries that perfuse tissues, such as the heart, kidneys, and lungs. However, many studies with passive targeted liposomes reported low selectivity that resulted in higher drug accumulation in healthy tissues and organs and low concentration of liposomes within tumor tissues, resulting in treatment failure. Furthermore, cationic charges were also reported to cause lower tumor penetration and non-specific accumulation, while neutral liposomes were able to penetrate deeper into tissue at the expense of a lower cellular uptake [12].
As an alternative, the active targeting of cancer sites uses various ligands to recognize antigens expressed by tumor cells (Figure 1). The targeting of cancer cells may be performed by antibodies or antibody fragments (immune-liposomes), aptamers, charged molecules, proteins, peptides, or other receptor-ligand bindings for site-specific targeting. Relying on the ligands, such as folic acid (FA), CD44 (cell surface glycoprotein), vascular endothelial growth factor (VEGF), integrin, etc., presented by a few types of tumors, the problem of tumor cell heterogeneity in the expression of their surface markers has not been overcome yet. A possible direction for future investigations is the ligand coupling of different internal and external stimuli for more sensitive detection. By attaching various chemical-specific moieties to the liposome’s surface, the system can respond to different physiology-dependent or physical stimuli. Stimuli-responsive liposomes are also considered to be effective in on-demand drug delivery. These “smart” nanocarriers undergo triggered drug release on physiology-dependent (internal) or external (physical such as light, temperature, magnetic field, ultrasound, etc.) stimuli, thus providing better accuracy in the concentration, timing, dosage, and location of release [13]. By modification of the vesicle surface, liposomes can be used both to impart “smart” character and attach ligands for active targeting. As shown in Figure 2, the active targeting of the drug-loaded carrier can respond to both extracellular and intracellular signals. The former dynamically target the liposomes toward the tumor tissue during circulation and trigger their accumulation, penetration, and internalization into the cancer cells [14]. The intercellular signals are responsible for the release of drugs in different cell compartments. Although these smart systems have been extensively explored as pharmacotherapy agents, different adverse effects have limited their clinical applications [15]. Therefore, the selection of proper constituents and activation mechanisms is a crucial factor in engineering the modified drug carriers, since this predetermines their distribution, targetability, and efficiency at specific sites.
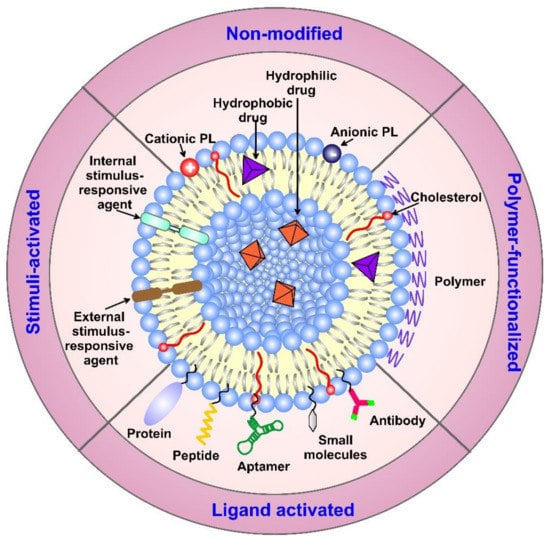
Figure 1. Surface modification strategies of liposomes, together with their classification. The modified carriers can contain active components, such as drugs, small molecules, proteins, and/or targeting moieties, such as antibodies, peptides, aptamers, etc., conjugated on the surface of the vehicles through different linkers, non-covalent or covalent bonds, and electrostatic interactions. Abbreviation: PL—phospholipid.
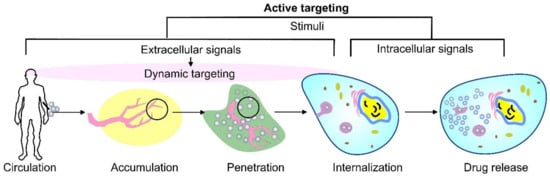
Figure 2. A schematic representation of cancer drug delivery by stimuli-responsive vehicles. Depending on the area of activation, the stimuli can be divided into extracellular signals that focus on dynamic targeting during circulation, accumulation, penetration, and internalization, as well as intracellular signals.
2. The State of the Art of Liposomes for Drug Delivery and their Production Methods
2.1. Main Characteristics of Liposomes for Drug Delivery
Liposomes, as nanomedicine representatives, have a rapidly evolving design that improves their interactions with cellular targets at the nanoscale. The functional scaffold for the fundamental cell-like kinetic behavior of liposomes that impart surface modification for active and passive targeting is the phospholipid bilayer envelope. Generally, the liposome drug-loaded vesicle consists of a hydrophilic interior part and shell of one or several concentric phospholipid bilayers or lipid monolayer structures, called micelles (Figure 3). Considering the water solubility of the loaded drug, the latter may be bonded to the surface of the vesicle, encapsulated in the aqueous core, or included in the hydrophobic space of the bilayer lipid membrane by Wan der Waals forces [16]. Due to the enhanced lipid–lipid exchange, the dissolution rate and convective flux of the entrapped drug are accelerated. Additionally, by modification of these drug-delivery vesicles, they may be targeted toward specific cells, tissues, or organs [17], while decreasing the systemic side effects.
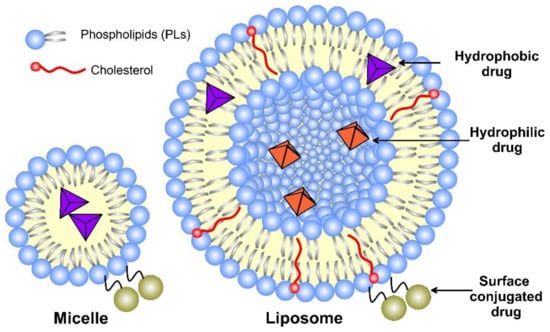
Figure 3. A schematic structure of micelle and unilamellar liposome, together with the possible drug location.
2.2. Factors That Influence the Physicochemical and Drug Delivery Properties of Liposomes
The physicochemical and drug delivery properties of liposomes depend on their composition, surface charge, size, number of lamellae, bilayer fluidity, surface modification for targeting, and production method [11]. These characteristics can be modified and uniquely tailored at a certain stage of the liposome’s production to favor specific biological, chemical, and mechanical properties and drug delivery targeting. The main factors affecting the stability of liposome formulation, their bioavailability, and drug delivery ability are as follows.
2.2.1. Bilayer Composition and Fluidity
There are several types of phospholipids used for the preparation of liposomes, such as natural phospholipids, modified phospholipids from natural sources, semi-synthetic or fully synthetic phospholipids, non-natural head group phospholipids, etc. Natural non-toxic phospholipids, sphingolipids, cholesterol, and hydrophilic polymers are typical constituents of the liposomal bilayer. The membranes of the liposomes used in medicine mostly consist of phosphatidylcholine (PC) and a little amount of phosphatidylethanolamine (PE) present within them both, with neutral charges under physiological pH [18]. Based on the overall charge of the lipid part of the vesicle, the liposomes can be anionic, cationic, or neutral.
The lipid composition and transition temperature (Tc) determine the curvature of liposomes. At Tc, phospholipids shift from gel to a liquid-crystalline phase with greater fluidity. Tc depends on the length of the fatty acid chains and their saturation [19]. Tc decreases with a decrease in chain length and an increase in the double bonds in them. The presence of unsaturated lipids within the liposomes compromises the integrity of the lipid bilayer through lipid transfer to lipoproteins, disintegration, and leakage of the content. Therefore, Tc predetermines the permeability and fluidity of the liposome bilayer. The fluidity of the bilayer is also affected by cholesterol, which increases the fluidity in the core of the bilayer because of its aromatic rings laying parallel to the fatty acid chains, while the viscosity is increased close to the phospholipid headgroups where its hydroxyl group is placed [20]. Cholesterol is used in different liposomal formulations because its presence stabilizes the carriers by protecting them from interactions with different proteins, such as transferrin, albumin, macroglobulin, etc. [21].
2.2.2. Lamellarity and Size
As amphiphilic structures, lipid materials and phospholipids spontaneously disperse in water to form physically stable liposomes, where the constituent lipids are usually not covalently bonded with each other, in contrast to the monomer units that build polymers. When consisting of only one phospholipid bilayer, the liposomes are termed unilamellar, while those containing several bilayers are called oligo- or multilamellar vesicles (Figure 4). Small unilamellar carriers, with sizes less than 100 nm, are usually smaller than multilamellar, but there are also large (>100 nm) and giant (>1000 nm) unilamellar vesicles. The amount of the loaded drug compound and the drug release rate are dependent on the number of phospholipid bilayers. Overall, the liposome size can vary between 20 nm to 2.5 μm, and both the size and number of bilayers determine the amount of the encapsulated drug [22]. As discussed, for injectable clinical applications, the liposome diameters should not exceed 200 nm, so they can be considered submicron or nanostructure carriers.
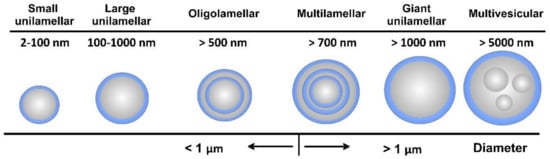
Figure 4.
Overall view of liposome types together with their indicative size.
2.2.3. Surface Charge
The surface charge of liposomes depends on the phospholipid head groups. Negatively charged phospholipids are faster recognized by macrophages than neutral phospholipids that shorten the blood circulation time. Neutral liposomes are stabilized by small negative charges, due to increasing the repulsive electrostatic forces affecting the aggregation-dependent mechanism of phagocytic uptake [11]. On the other hand, cationic liposomes undergo opsonization (interaction with plasma proteins) that triggers phagocytic-mediated clearance by the liver, spleen, and lung. Because the uptake of positively charged liposomes appeared to be higher than that of negatively charged, most of the FDA-approved liposomes are negatively charged [19]. Moreover, cationic liposomes also hinder interactions with tumor cells, and their accumulation in tumor stroma just performs the function of a drug-loaded depot.
2.2.4. Stability and Bioavailability
To predict the bio-behavior of a potential nanocarrier in the body, the protein adsorption (the so-called protein corona) on the liposomal surface should be considered. Since changing the liposomal biological identity, the absorbed proteins determine the organism’s response, including the cell uptake body distribution and clearance [23]. Even liposomes that are synthesized from natural phospholipids are recognized as foreign particles in the body and cleaned by the mononuclear phagocyte system [24]. By choosing either natural or synthetic (phospho)lipids as ingredients, the lifespan, biocompatibility, and biodegradability of a liposome could be changed.
The first (conventional) generation of liposomes that load drug molecules to their unaltered surface faces (Figure 3) challenges, due to its inherited instability. Their major shortcoming includes the lack of fast and easy preparation routes, rapid decomposition in the organism before achieving the therapeutic effect, low degree of drug-loading capacity, and instability in the bio-environment [25]. The next-generation liposome formulations overcame the tendency to fuse because of high surface tension and escaped unspecific plasma protein adsorption by coating them with polymers, such as PEG (called “stealth liposomes” with a size less than 200 nm) [26] or super-hydrophilic zwitterionic polymers [27]. Hydrophobic long-chain polymers, such as PEG and glycolipids, are known to prevent rapid clearance and increase blood circulation time [28]. Liposome encapsulation reduces drug clearance by the immune and renal systems and increases their availability in the organism [29]. It was found that PEGylated small-sized (100–150 nm diameter) liposomes showed fewer interactions with opsonin [30], thus reducing their consumption by the reticuloendothelial system. The long-circulating “stealth” liposomes are found to target the cancer cells by the EPR mechanism, thus decreasing the drug toxicity in the organism. PEG encapsulation was successfully proven in the FDA-approved nanomedicine Doxil® [29]. Additionally, a variety of affinity ligands, such as peptides and antibodies, can be immobilized to the liposome formulations with PEG linkers for targeting the disease cell [31].
Hybrid liposomes consisting of a solid organic or metal oxide core and lipid shell and characterized by good size, morphology, mechanical stability, and drug-release kinetics have been proposed [32]. The lipid shell reduces drug diffusion, limits water penetration across the interface, and mimics the biological membrane. Recently, for stability enhancement and suitable drug encapsulation, solid lipid NPs, instead of liquid lipids, in the preparation method have been proposed. Such NPs demonstrated biocompatibility, biodegradability, acceptable bioavailability, higher shell life, improved drug targeting release, absorption, and dissolution, as well as easy large-scale production and sterilization [33]. However, the loading capacity of hydrophilic drugs is limited. Higher loading capacity, with a wider range of drugs, was obtained in the solid and liquid phases of lipids with imperfect crystalline structures [34]. When hydrophilic drugs are loaded by a covalent bond to hydrophobic molecules, resulting in the formation of salt, the lipid-drug conjugate can protect sensitive drugs from the acidic stomach conditions, while a polymer (such as PCL and PLGA) –liquid hybrid NP can form a core-shell structure when conjugating with drugs [35]. When delivering lipophilic drugs, the latter is maintained in its solubilized form in the lipids and by using lipid excipients, such as triglycerides, mixed glycerides, polar oils, surfactants, and co-solvents, and various favorable reactions, such as improved bioavailability, antioxidant effect, topical delivery, enhanced drug therapeutic effect, etc., are observed [36]. In developing such lipidic products for poorly aqueous soluble drugs, the issues related to difficulties in the manipulation and weak stability of lipid formulations can be successfully unraveled. To further enhance the therapeutic efficacy, liposomes with stimuli-responsive drug release have been developed.
2.3. Production Routs of Liposomes
Assembly methods play an essential role in liposome characteristics, including drug encapsulation efficiency and drug release profiles. Both “bottom-up” and “top-down” engineering approaches have been used to form individual small vesicles (Figure 5). Numerous synthetic liposomes-like DDS with finely tuned physicochemical properties were synthesized through “bottom-up” processes, but it is still hard to achieve complex functionalities.
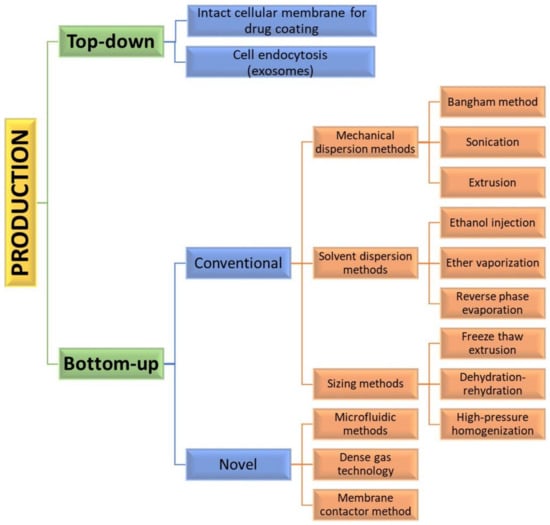
Figure 5.
Diagram of various manufacturing engineering approaches and processes used for the synthesis of liposome vesicles.
Natural cellular membrane-derived vehicles made by “top-down” techniques inherit their natural functionalities, or even enhance them, by using genetic modification. Similar to the mammalian exosomes that are released in the extracellular space, exosome-bound tumor antigens were found to induce a more active antigen-specific antitumor response than the corresponding soluble antigens [37]. When the membrane phospholipids are disturbed, they tend to rearrange into small spherical particles composed either of monolayers (micelles) or bilayers (liposomes). Mesenchymal stem cells (MSC) capable of homing to different cancer cells are often used as a source for producing anticancer-targeting vesicles [38]. However, the formation of liposomes is not a spontaneous process.
The encapsulation of synthetic nanoparticles with cellular membrane can be achieved by their internalization by cell endocytosis and the subsequent release of the vesicle-enclosed particle (exosome formation) [39] or by collecting intact cellular membranes that are afterward used for coating inert or biodegradable particles. Direct loading of exosomes incubated with a certain compound or simple drug mixing with the exosomes has been also reported [40]. For using exosomal carriers in drug delivery, it may be necessary that the exosomal interface be modified by fusing synthetic liposomes and exosomes. In that way, immunogenicity is decreased, while the colloidal stability and half-life of exosomes in the blood are improved [41]. This separate preparation of particles and top-down approach for preparing cellular membranes offers good flexibility and biological stability [42]. The use of natural membranes saves labor-intensive processes, such as protein identification, purification, and conjunction. However, adverse effects on cargoes and liposomes, such as aggregation, could also be expected.
The “bottom-up” strategies for liposome preparation can be classified into three main groups: mechanical dispersion methods, solvent dispersion methods, and size-adjusting methods. All of them use the precipitation of dissolved lipids into an aqueous solution and because of changed solubility, liposome formations occur spontaneously. The mechanical methods include the Bangham method, which produces liposome formations via thin lipid films deposited by organic solution on the glass surface by shaking at temperatures higher than Tc [43]. After that, the solvent is removed, and the lipid film is hydrated, while agitating to the lamellas from the surface to form spherical structures with heterogenous micron sizes. Another mechanical approach is sonication under a passive atmosphere with a bath or a probe sonicator to obtain liposome carriers with diameters down to 15–25 nm [44]. By applying dual asymmetric centrifugation, the mechanical turbulence and cavitation produce nanoliposomes with a size of around 60 nm and homogenous size distribution but poor productivity [45]. A third mechanical dispersion method is membrane extrusion, which consists of extrusion above the phase transition temperature through polycarbonate pore-containing membranes, allowing the formation of liposomes with dimensions close to that of the membrane pores’ size. The method is simple and reproducible in downsizing, but sensitive to product losses [46].
The solvent dispersion methods include ether vaporization and ethanol injection routes. The former consists of slow ether injection to a mixture of lipids into a warm aqueous solution. As a result of ether removal under vacuum and heat, unilamellar carriers with good size distribution and higher volume trapping activity liposomes are formed. During the ethanol injection, the lipids dissolved in the organic phase are injected into aqueous media, thus forming liposomes. However, since some liposomes are poorly soluble in ethanol, adequate mixing is not achieved. Additionally, the liposome population is heterogeneous, while alcohol removal is difficult. The presence of residual solvents in the bilayer can change the physical and mechanical characteristics of the membrane [47]. However, solvent injection methods are suitable to become continuous production routes. The alternative solvent dispersion method is reverse-phase evaporation, where different phospholipids and cholesterol can be used. The lipids are dissolved in an organic solvent, where inverted micelles are produced and shaped by sonication in a mixture of a buffered aqueous solution. The water-soluble molecules are encapsulated into liposomes, and the slow elimination of the organic solvent converts the micelles from viscous to gel form. The aqueous volume-to-liquid ratio in these formulations is high, making them suitable for entrapping a large percentage of aqueous material. However, the encapsulated compound is in contact with the organic solvent and has brief sonication periods, which makes the process unsuitable for fragile molecules, such as peptides or DNA strains [26].
The sizing methods include freeze-thaw extrusion, which creates large unilamellar vesicles, due to the fusion of small unilamellar liposomes during repeating cycles of freeze-thaw and vortexing the sample. Similarly, during the dehydration–rehydration technique, small unilamellar liposomes in the buffer are mixed with the moiety to be entrapped and then freeze-dried. After rehydrating the vesicles, larger formulations are constructed because the frozen phase becomes more concentrated and flattened. Since heterogeneity of the size is observed, sizing by sequential extrusion at low pressure through polycarbonate membranes or gel-permeation chromatography [48] can be applied. The size reduction follows the mechanism of rupture at the entrance of the membrane pore and rearrangement during the membrane passage. During the high-pressure homogenization technique, the liposome suspension is passed through a narrow gap under high pressure and broken down by the cavitation, turbulence, and shear force of the velocity gradient and, after that, re-arranged into smaller liposomes. By increasing the pressure and process cycles, the polydispersity and particle size decrease, which results in decreased encapsulation efficiency [49].
All these production routes are characterized by some disadvantages, such as the need for a large amount of organic solvent, poor drug loading efficacy, low yield, and time-consuming issues [50]. Bulk methods also produce products that are not uniform in size and lamellarity due to poor control over the chemical and mechanical conditions of the process. Additionally, these techniques may not be suitable for processing various biomolecules that can undergo structural changes [50].
By applying precise control of fluids in a constrained volume, novel microfluidic methods offer the ability to remove the organic phase from the final product, a high degree of control over the production route, and reproducibility in the production of monodisperse vesicles [51]. By using microfluidic systems, many factors, such as osmolarity, pH, temperature, vesicle size, salinity, and fluid mechanical forces, can be precisely controlled. Such methods are pulse jetting [52], ice droplet hydration [53], hydrodynamic focusing [54], hydrodynamic pinch-off mechanism [55], solvent extraction-based droplet microfluidics [56], etc. The main disadvantages of microfluidic systems include the use of low quantities of solution, resulting in a low volume of the manufacturing process and rather clumsy methods for establishing and operating. Another new method of liposome formation namely dense gas technology, employs supercritical fluids, such as supercritical carbon dioxide, that are excellent solvents for many lipids; after mixing with the water phase, liposomes with narrow size distribution are synthesized [57]. The next modern production route is the membrane contactor method, in which a lipid phase dissolved in alcohol is pushed through a porous membrane into an aqueous phase flow, where lipid molecules are self-assembled into homogenous-size liposomes [58]. All these modern methods have high scaling-up abilities, allowing for large-scale liposome production, but until now, the disadvantages of these novel techniques were mainly connected with their high capital cost [50], which circumstances can discourage their industrial development. Furthermore, strict control over quality, purity, on-shelf stability, and sterility is required by pharmaceutical regulations, which can represent a limit to efficient technology transfer.
2.4. Drug Encapsulation Techniques
Methods for encapsulating different drug agents within liposomes are either passive when the cargo is encapsulated during liposome formation or active when the loading follows the formation of empty liposomes. Hydrophobic drugs can be directly combined into liposome formations during carrier formation, and the trapping effectiveness depends on the solubility of the drug in the liposomal membrane and may reach 100% [26]. The passive encapsulation efficacy depends on the aqueous volume enclosed by the vesicle, which is proportional to phospholipid concentration in the dispersion and morphology of the vesicle.
Water-soluble drugs are usually actively entrapped by employing, for example, the blending of empty liposomes with a concentrated drug solution that distributes equally by diffusion [59]. The method that creates diffusion gradients is called “remote loading”. For increasing the loading effectiveness, pH gradients across the bilayer or ion gradients can be used [60]. Transmembrane proton gradient can be generated by preparing liposomes in low pH buffers or by incorporating ionophores that couple the outward movements of mono or divalent cations with the inward movement of protons, thus acidifying the liposome interior. Another route is the preparation of liposomes in the presence of a weak base, such as ammonium sulfate. The removal of the external ammonium salt generates a pH gradient that helps the drug-loading process. When loading two different drugs in the same liposome system, a combination of passive and active encapsulation could be applied. For example, cytarabine is passively loaded into the liposomes when hydrating the lipid foams and after sizing, and a daunorubicin buffer solution is incubated with the cytarabine-loaded liposomes. Daunorubicin diffuses through the lipid bilayer and is actively accumulated inside the liposome, due to the copper gluconate/triethanolamine-based loading [61].
An efficient strategy for loading drugs within liposomes is covalent linkage. For example, muramyl tripeptide-phosphatidyl ethanolamine (MTP-PE) was linked with a peptide spacer—a formulation that had an improved lipid solubility, rather than muramyl dipeptide itself (a component of the cell wall of Gram-positive bacteria), has been used for loading drugs within liposomes, such as in Mepact. These amphiphilic molecules were able to intercalate into the phospholipid membrane during liposome synthesis, and no free MTP-PE existed [62].