Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Stefano Menini and Version 2 by Peter Tang.
Oxidative stress and hypoxia-inducible factors (HIFs) have been implicated in the pathogenesis of diabetic cardiovascular and renal diseases. Reactive oxygen species (ROS) mediate physiological and pathophysiological processes, being involved in the modulation of cell signaling, differentiation, and survival, but also in cyto- and genotoxic damage. As master regulators of glycolytic metabolism and oxygen homeostasis, HIFs have been largely studied for their role in cell survival in hypoxic conditions.
- advanced glycation end products
- atherosclerosis
- diabetic kidney disease
- inflammation
- methylglyoxal
- prolyl hydroxylase domain proteins
- reactive oxygen species
- sirtuin-1
- sodium glucose co-transporter 2 inhibitors
1. Introduction
Diabetes represents a growing global health issue that significantly contributes to premature mortality, morbidity, and disability, and poses a tremendous economic burden to national health systems [1][2][1,2]. Most of the harms and costs related to diabetes are due to its long-term microvascular (nephropathy, retinopathy, and neuropathy) and macrovascular (coronary, cerebrovascular, and peripheral artery disease) complications [3]. Redox imbalance, dysregulated hypoxia-inducible factor (HIF) signaling, and mitochondrial abnormalities have all been implicated in vascular tissue damage associated with diabetes.
The role of reactive oxygen species (ROS) in diabetic complications has been recently reviewed [4][5][4,5]. Hyperglycemia and other diabetes-related metabolic abnormalities cause ROS overproduction in endothelial cells and perivascular cells of large and small vessels [6]. Although oxidative metabolism in mitochondria has long been considered the main source of superoxide overproduction in the setting of hyperglycemia [7], other cytosolic and plasma membrane oxidoreductases, such as NAD(P)H oxidases (NOXs), cycloxygenase-2, myeloperoxidase, etc., provide a substantial contribution to ROS production in diabetic tissues [4]. In addition to hyperglycemia, several other diabetes-associated stimuli may promote ROS formation and redox abnormalities, including accumulation of advanced glycation end products (AGEs) and their carbonyl precursors, inflammation, dyslipidemia, and upregulation of the renin-angiotensin system (RAS) [4][5][4,5]. Hence, oxidative stress is considered a key contributor to the pathogenesis of diabetes-related end-organ damage [8][9][10][8,9,10]. According to these premises, antioxidants might be predicted to protect against diabetic vascular complications. In fact, antioxidant therapies have failed to provide substantial clinical benefit [11]. This paradox might be explained by recent findings that, besides representing an important pathomechanism, oxidative stress and ROS are also part of essential signaling networks modulating different cellular functions, such as energetic metabolism, growth, survival, angiogenesis, vascular tone regulation, etc. [4]. Consistent with a regulatory function in fundamental cellular processes, physiological enzymatic sources of ROS such as NOXs are evolutionarily conserved enzymes whose only identified function is to produce ROS [11]. In this perspective, ROS are not merely toxic and metabolic waste products that need to be removed to prevent cellular damage. The final result of ROS action does not depend exclusively on their absolute concentration (i.e., the so-called “redox balance hypothesis”), but also on the amount and type of ROS produced in specific subcellular compartments and, above all, on their interactions with pathways and systems that are closely related to energy and oxygen metabolism.
HIFs are master regulators of oxygen homeostasis, playing a key role in the adaptive regulation of energy metabolism in mammalian tissues [12]. The HIF pathway has been implicated in the development of cardiovascular and renal complications of diabetes [13] (Figure 1).
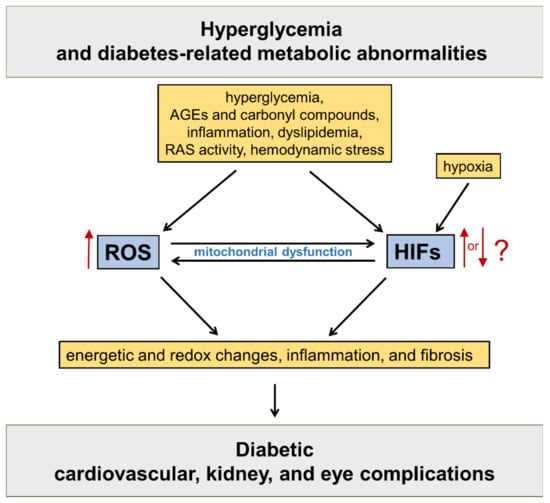
Figure 1. Oxidative stress and hypoxia-inducible factors (HIFs) in the pathogenesis of diabetic vascular complications. Refer to the main text for detailed description. AGEs = advanced glycation end products; RAS = renin angiotensin system; ROS = reactive oxygen species.
2. The HIFs Family
HIFs are heterodimeric transcription factors acting as master regulators of oxygen homeostasis. By regulating a large battery of genes and activating a broad range of transcriptional responses, HIFs match oxygen supply and demand in tissues [12] (Figure 2).
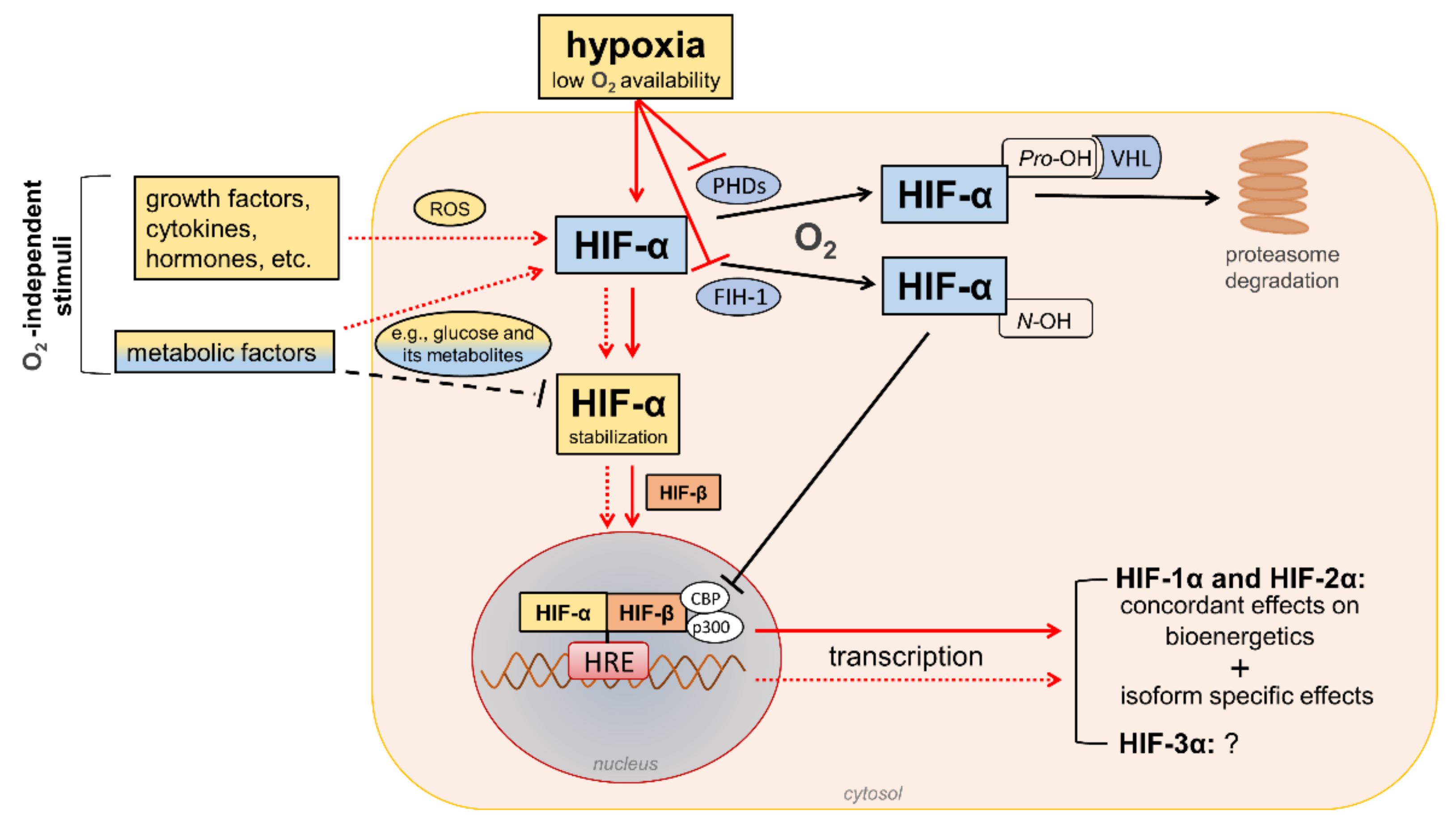
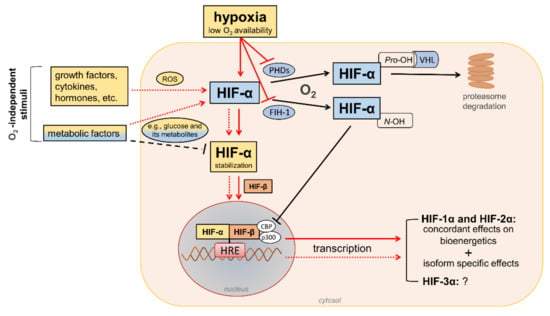
Figure 2. Regulation of hypoxia-inducible factor (HIF) signaling by oxygen (O2)-dependent and O2-independent mechanisms. There are conflicting opinions on the effects of metabolic factors, particularly hyperglycemia, on the stability of HIF-α proteins (mainly HIF-1α). Refer to the main text for detailed description. Black arrows/lines and light blue boxes/ovals indicate components and processes of the HIF-α degradation pathway in oxygenated cells (black solid arrows/lines) or metabolic factors that may impair HIF-α stabilization in hypoxic conditions (black dashed line). Red arrows/lines and yellow boxes indicate oxygen-dependent (red solid arrows/lines) and oxygen-independent stimuli (red dotted lines) promoting the stabilization and transcriptional activity of HIF-α proteins. ROS = reactive oxygen species; PHDs = prolyl hydroxylase domain proteins; FIH-1 = factor-inhibiting HIF-1; Pro-OH = proline hydroxylation; N-OH = asparagine hydroxylation; VHL = von Hippel–Lindau; CBP = cAMP response element-binding protein; p300 = E1A binding protein p300; HRE = hypoxia-response element.
The HIF family includes three hypoxia-regulated α-subunits (HIF-1α, HIF-2α, and HIF-3α), which are associated with a constitutively expressed β-subunit (HIF-1β). HIF-1α and HIF-2α are the principal HIF-α isoforms. Despite an identical consensus recognition sequence in DNA, HIF-1α and HIF-2α do not compete for binding sites and activate common as well as unique target genes [14][15][28,29]. Therefore, HIF-1α and HIF-2α may function more independently than previously thought, suggesting the need for isoform-specific HIF inhibitors for specific therapeutic indications [16][30]. Little is known about HIF-3α. This isoform has been considered a negative regulator of the hypoxic response by competing with the other two isoforms [17][31], but its role has yet to be fully defined.
In the presence of oxygen, iron, and 2-oxo-glutarate (α-ketoglutarate), the HIF-α subunits are continuously targeted for proteasomal degradation by three HIF prolyl hydroxylase domain proteins (PHD1-3). PHD2 plays a prominent role in the regulation of the HIF signaling pathway [18][19][32,33] as it is the rate-limiting enzyme for the degradation of the HIF-1α subunit in normoxia [18][32]. PHDs are α-ketoglutarate—dependent dioxygenases that rapidly hydroxylate HIF-α subunits at two proline residues (Pro-402 and Pro-564 in HIF-1α) in the presence of oxygen. Prolyl hydroxylated HIF-α subunits are recognized by the von Hippel–Lindau (VHL), a ubiquitin E3 ligase complex that marks HIF-α proteins for proteasomal degradation [19][33]. In addition to the negative regulation of protein stability by PHDs, the transactivation function of HIF-1α and HIF-2α is inhibited by factor-inhibiting HIF-1 (FIH-1) [20][34], another oxygen sensor that hydroxylates HIF-α on an asparagine residue (N-803) in its C-terminal transactivation domain [21][35]. This oxygen-dependent post-translational modification prevents the interaction of HIF-α proteins with transcriptional co-activators, such as cAMP response element-binding protein (CBP) and E1A binding protein p300 [22][23][36,37]. Therefore, PHD and FIH enzymes ensure the full repression of the HIF pathway in normoxia by controlling the degradation and transcriptional activity, respectively. Conversely, insufficient levels of oxygen (i.e., hypoxia) inhibit hydroxylation, impair HIF-α proteasome degradation, and favor HIF-α stability and dimerization with the HIF-1β subunit, which facilitates translocation to the nucleus, the recruitment of transcriptional coregulators, and binding to a hypoxia response element (HRE) in various target genes. Overall, oxygen-dependent hydroxylations on proline and asparagine residues modulate HIF-α stability and activity so that changes in oxygen availability are transduced to the nucleus as changes in HIF-dependent transcriptional activity.
In addition to hypoxia, HIF-α proteins respond to oxygen-independent stimuli, including growth factors, cytokines, vasoactive peptides, coagulation factors, and hormones such as insulin [24][25][26][27][28][38,39,40,41,42]. Many metabolic factors, including glucose and their glycolytic metabolites, can also affect the HIF pathway in opposite directions [29][30][43,44], possibly depending on the availability of oxygen. The functions of HIFs in oxygenated cells and the mechanisms involved in the regulation of HIF signaling by metabolic stimuli in normoxic or hypoxic conditions are not yet fully understood. Nevertheless, the modulation of HIF activity by metabolic changes suggests its contribution in the reactive adaptation to dynamic microenvironment variations, and also in the absence of alterations in oxygen tension. Regarding the mechanism(s), most of the nonhypoxic stimuli capable of modifying HIF activity induce ROS production as part of their signaling cascade [31][45]. Therefore, a role of ROS in HIF-α stabilization and activity has been hypothesized, particularly under normoxia conditions.
3. Redox Regulation of HIFs: Role of Diabetes-Related Stimuli
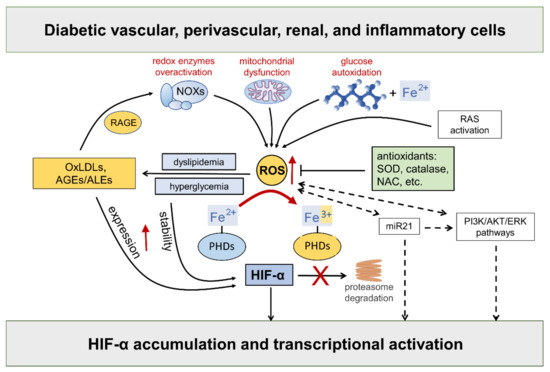
Hyperglycemia-driven mitochondrial dysfunction [32][46], the over activation of redox-enzymes in vascular and inflammatory cells [4], and glucose autoxidation catalyzed by trace amounts of transition metal ions [33][47], such as iron and copper, are deemed the major source of ROS in diabetic vascular tissues. In addition, as part of the cardiometabolic syndrome, the activation of the RAS system triggers ROS formation via angiotensin (Ang) II type 1 receptor stimulation, thus contributing to endothelial dysfunction and diabetes-associated cardiorenal diseases [34][48]. Compelling evidence exists that ROS take part in the regulation of HIF-α [35][36][37][38][39][25,49,50,51,52] (Figure 3).
Figure 3. Hypoxia-inducible factor (HIF)-α stability is sensitive to redox status in normoxia: role of diabetes-related stimuli. Refer to the main text for detailed description and references. Dashed arrows = regulation of HIF-α by ROS via indirect mechanisms. NOXs = NADPH oxidases; RAGE = receptor for AGEs; RAS = renin angiotensin system; AGEs = advanced glycation end products; ALEs = advanced lipoxidation end products; OxLDLs = oxidized Low-Density Lipoprotein; ROS = reactive oxygen species; SOD = superoxide dismutase; NAC = N-acetyl-L-cysteine; PHDs = prolyl hydroxylase domain enzymes; miR21 = microRNA 21; PI3K = phosphoinositide 3-kinase; Akt = Ak strain transforming; ERK = extracellular signal-regulated kinase; PHDs = prolyl hydroxylase domain enzymes.
While the role of mitochondrial ROS under hypoxia has long been debated with no clear consensus, there is considerable evidence in support of the normoxic stabilization of HIF-α proteins by ROS. The first demonstration that oxidative stress contributes to HIF-α stabilization under normoxia comes from studies showing that the addition of exogenous ROS, or ROS-generating enzymes, is sufficient to stabilize HIF-1α protein and induce the transcription of HIF target genes, such as vascular endothelial growth factor (VEGF) [36][38][40][49,51,53]. Moreover, all these effects were attenuated by cellular treatments with antioxidant compounds such as catalase, glutathione, N-acetyl cysteine, and vitamins E and C [41][42][43][44][45][46][47][48][49][50][51][54,55,56,57,58,59,60,61,62,63,64]. HIF-2α stability is similarly affected by the addition of ROS [40][53], a finding confirmed and extended by other studies showing that NOX4-derived hydrogen peroxide (H2O2) increases HIF-2α stability and its transcriptional activity [51][52][26,64]. Approaches directed at increasing the levels of the superoxide anion by silencing or the pharmacological inhibition of superoxide dismutase (SOD) also demonstrated a role for ROS in HIF-1α expression and protein accumulation in normoxia [53][54][65,66], but not in hypoxia [53][65]. Overall, these findings implicate ROS in normoxic HIF-α stabilization and activity by showing that HIF-α stability is sensitive to the redox status in many cell types and that ROS such as superoxide and H2O2 are sufficient for activating the HIF pathway.
As far as the mechanism, ROS were shown to inhibit PHD activity and VHL binding to HIF-1α under normoxia [53][55][65,67]. It has been suggested that ROS may oxidize the iron co-factor of the PHD enzymes, thus inhibiting their activity [56][68]. Hence, sustained ROS production may reduce the cellular pool of ferrous ion, which is required for the full activity of PHD enzymes [57][58][69,70]. Consistently, adding cobalt ions to cells with normal oxygen tension mimics a hypoxia response through a competition mechanism between cobalt and ferrous ions for binding with the enzyme active site, thus impairing PHDs’ activity and HIF-α hydroxylation [59][71]. Likewise, the iron chelator desferrioxamine causes HIF-α proteins to accumulate in a dose-dependent manner [60][61][62][72,73,74].
In addition to affecting HIF-α protein levels by the disruption of PHD and/or FIH-1 activity, ROS have also been suggested to mediate the transcriptional and translational regulation of HIF-α via indirect mechanisms, including phosphoinositide 3-kinase (PI3K)-Ak strain transforming (Akt) and extracellular signal-regulated kinase (ERK) pathways. A key role of ERK and PI3K/Akt signaling in ROS-mediated HIF-1α stabilization has been suggested in many disease conditions including neoplastic, ischemic, and inflammatory disorders [35][63][25,75]. Consistently, tyrosine kinase inhibitors were demonstrated to block HIF-α synthesis and activity in hypoxia, showing that protein phosphorylation plays an important role in HIF signaling [35][63][25,75]. In addition, many nonhypoxic stimuli, including insulin, thrombin, and growth factors were shown to stimulate the HIF response through ERK and PI3K/Akt signaling pathways in a redox-sensitive manner [25][41][43][64][65][66][67][68][69][70][71][39,54,56,76,77,78,79,80,81,82,83].
In diabetes, excessive ROS generation triggers the production of proinflammatory compounds, such as oxidized LDLs (oxLDLs) and advanced glycoxidation/lipoxidation end products (AGEs/ALEs), which may serve as additional nonhypoxic stimuli for HIF activation. The levels of these byproducts of glucose and lipid peroxidation are increased in the bloodstream and tissues of diabetic subjects, where they propagate metabolic and redox signals through interactions with several specific receptors, in particular the receptor for AGEs (RAGE) [72][73][74][84,85,86]. RAGE signaling elicits the activation of multiple intracellular signaling pathways, including the protein kinases Akt, ERK1/2 mitogen activated kinases, and c-Jun N-terminal kinase. Eventually, these signaling pathways result in the activation of redox-sensitive and proinflammatory transcription factors such as nuclear factor kappa B (NF-κB) and activator protein 1 (AP-1) [72][73][74][75][84,85,86,87]. Importantly, oxLDLs and AGEs/ALEs have been involved in the enhancement of HIF-α protein expression [76][77][88,89], stability [45][77][78][58,89,90], and transcriptional activity [79][80][81][91,92,93] in diabetic vasculopathy. Both hyperglycemia and AGEs/ALEs promote the protein accumulation of HIF-α proteins—mainly HIF-1α—and HIF activity in glomerular mesangial and renal tubular epithelial cells in in vitro and in vivo models of DKD [82][83][17,94]. Moreover, the AGE-RAGE signaling pathway augments ROS generation by NOX enzymes [84][95], thereby generating a vicious cycle that boosts oxidative stress, AGE/ALE production, and HIF signaling dysregulation. Altogether, these findings support the hypothesis of a role for the redox regulation of HIF activity by diabetes-related, nonhypoxic stimuli in the pathogenesis of cardiovascular and renal complications of diabetes.
Finally, a potential role for some microRNAs (miR) in the redox- and PI3K/Akt-dependent stabilization of HIF-1α has been proposed [35][25]. In particular, miR21 is induced by ROS [85][96] and plays a role in both PI3K-Akt and HIF-1α signaling activation [86][97]. Interestingly, miR21 has also been implicated in diabetes development and ROS-mediated damage [87][98] as well as DKD [88][89][99,100]. Together with the observation that PI3K and Akt signaling is altered by hyperglycemia [90][101], these findings suggest the involvement of a miR21-PI3K-Akt axis in the redox regulation of HIF in diabetic complications.