Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Kok Yong Chin.
Osteoporosis refers to excessive bone loss as reflected by the deterioration of bone mass and microarchitecture, which compromises bone strength. It is a complex multifactorial endocrine disease. Its pathogenesis relies on the presence of several endogenous and exogenous risk factors, which skew the physiological bone remodelling to a more catabolic process that results in net bone loss.
- bone
- menopause
- osteoblast
- osteoclast
1. Introduction
The world has been experiencing an increase in lifespan due to improved medical care and living environment, but this has not kept pace with the increase in healthy life expectancy [1]. Ageing causes multiple adverse physiological changes to the body due to the lifetime accumulation of molecular and cellular damage [2]. Among these geriatric diseases, the ageing of the skeleton is one aspect often overlooked by the community and medical professionals alike. Generally, humans achieve peak bone mass in the third decade of life, but the exact age varies with sex and skeletal sites [3]. After peaking, both sexes experience a decline in bone mass [4], which is accelerated during menopause in women [5].
Osteoporosis is a skeletal disease characterised by reduced bone strength due to deteriorating bone mass and bone microarchitecture, leading to increased susceptibility to fracture [6]. Owing to a lower peak bone mass and faster bone loss during menopause, women are at greater risk for osteoporosis than men [5]. While the development of osteoporosis is mostly asymptomatic, its ultimate consequences, i.e., fragility fractures, pose tremendous medical and economical challenges to the patients and society [7]. Despite the availability of effective therapy, a substantial number of patients with osteoporosis remain untreated [8].
2. Pathophysiology of Osteoporosis
The traditional pathophysiological models of osteoporosis are based on endocrine mechanisms. Two examples are estrogen deficiency in postmenopausal women and secondary hyperparathyroidism in the elderly due to menopause and vitamin D deficiency. In reality, osteoporosis is a multifactorial disease caused by a complex interplay of genetic, intrinsic, exogenous, and lifestyle factors [9]. A basic understanding of the bone remodelling cycle will facilitate the discussion on the pathophysiology of osteoporosis (Figure 1). Osteoclasts, osteoblasts, and osteocytes are the three main players in bone remodelling. When bone damage occurs, the macrophage polykaryon-derived osteoclasts migrate to the damage site and perform bone resorption [10]. At the end of bone resorption, osteoclasts undergo apoptosis and produce apoptotic bodies that may play a role in the subsequent osteogenesis [10]. After the reversal phase, the mesenchymal stem cell-derived osteoblasts will migrate to the cavity and perform bone formation [11]. Some osteoblasts will be embedded in the bone matrix they synthesise and differentiate into osteocytes. Osteocytes act as a mechanosensor and play regulatory roles in regulating the bone remodelling process through signalling proteins and via perilacunar remodelling directly [12].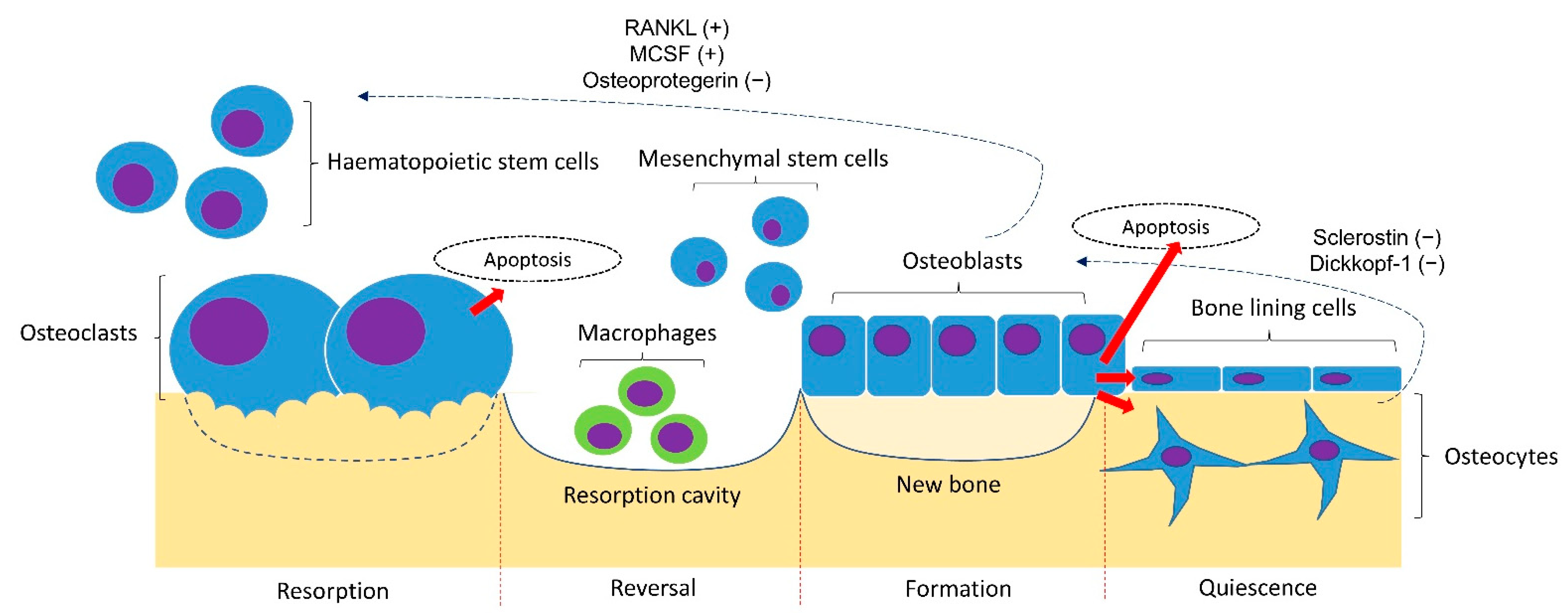
Figure 1. Bone remodelling cycle. The bone remodelling cycle is governed by osteoclasts, osteoblasts and osteocytes derived from the respective stem cell lineage. The differentiation of osteoclasts is stimulated by the receptor activator of nuclear factor kappa-B ligand (RANKL) and macrophage colony-stimulating factor (MSCF) and inhibited by osteoprotegerin (OPG) synthesised by osteoblasts and osteocytes. The osteogenesis of osteoblasts is inhibited by sclerostin and Dickkopf-1 synthesised by osteoblasts. Notes: +, promoting factor; −, inhibiting factor.
References
- World Health Organisation. GHE: Life Expectancy and Healthy Life Expectancy. Available online: https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-life-expectancy-and-healthy-life-expectancy (accessed on 28 September 2022).
- Niccoli, T.; Partridge, L. Ageing as a Risk Factor for Disease. Curr. Biol. 2012, 22, R741–R752.
- Berger, C.; Goltzman, D.; Langsetmo, L.; Joseph, L.; Jackson, S.; Kreiger, N.; Tenenhouse, A.; Davison, K.S.; Josse, R.G.; Prior, J.C.; et al. Peak bone mass from longitudinal data: Implications for the prevalence, pathophysiology, and diagnosis of osteoporosis. J. Bone Miner. Res. 2010, 25, 1948–1957.
- Specker, B.L.; Wey, H.E.; Smith, E.P. Rates of bone loss in young adult males. Int. J. Clin. Rheumtol. 2010, 5, 215–228.
- Alswat, K.A. Gender Disparities in Osteoporosis. J. Clin. Med. Res. 2017, 9, 382–387.
- Lorentzon, M.; Cummings, S.R. Osteoporosis: The evolution of a diagnosis. J. Intern. Med. 2015, 277, 650–661.
- Mitchell, P.J.; Chan, D.D.; Lee, J.K.; Tabu, I.; Alpuerto, B.B. The global burden of fragility fractures—what are the differences, and where are the gaps. Best Pract. Res. Clin. Rheumatol. 2022, 101777.
- Hachuła, M.; Pietrzyk, B.; Gruszka, W.; Cedrych, I.; Chudek, J. High rates of undiagnosed and untreated osteoporosis in postmenopausal women receiving medical services in the area of Upper Silesia. Prz. Menopauzalny 2020, 19, 72–79.
- Clarke, B.L.; Khosla, S. Physiology of bone loss. Radiol. Clin. N. Am. 2010, 48, 483–495.
- Ma, Q.; Liang, M.; Wu, Y.; Luo, F.; Ma, Z.; Dong, S.; Xu, J.; Dou, C. Osteoclast-derived apoptotic bodies couple bone resorption and formation in bone remodeling. Bone Res. 2021, 9, 5.
- Xiao, W.; Wang, Y.; Pacios, S.; Li, S.; Graves, D.T. Cellular and Molecular Aspects of Bone Remodeling. Front. Oral Biol. 2016, 18, 9–16.
- Prideaux, M.; Findlay, D.M.; Atkins, G.J. Osteocytes: The master cells in bone remodelling. Curr. Opin. Pharmacol 2016, 28, 24–30.
- Tobeiha, M.; Moghadasian, M.H.; Amin, N.; Jafarnejad, S. RANKL/RANK/OPG Pathway: A Mechanism Involved in Exercise-Induced Bone Remodeling. Biomed. Res. Int. 2020, 2020, 6910312.
- Chin, K.Y.; Ekeuku, S.O.; Pang, K.L. Sclerostin in the development of osteoarthritis: A mini review. Malays J. Pathol 2022, 44, 1–18.
- Siddiqui, J.A.; Partridge, N.C. Physiological Bone Remodeling: Systemic Regulation and Growth Factor Involvement. Physiology 2016, 31, 233–245.
- Ji, M.X.; Yu, Q. Primary osteoporosis in postmenopausal women. Chronic Dis. Transl Med. 2015, 1, 9–13.
- Chin, K.Y. The Relationship between Follicle-stimulating Hormone and Bone Health: Alternative Explanation for Bone Loss beyond Oestrogen? Int. J. Med. Sci. 2018, 15, 1373–1383.
- Chin, K.Y.; Ima-Nirwana, S. Sex steroids and bone health status in men. Int. J. Endocrinol. 2012, 2012, 208719.
- Mohamad, N.V.; Soelaiman, I.N.; Chin, K.Y. A concise review of testosterone and bone health. Clin. Interv. Aging 2016, 11, 1317–1324.
- Khosla, S.; Oursler, M.J.; Monroe, D.G. Estrogen and the skeleton. Trends Endocrinol. Metab. 2012, 23, 576–581.
- Mohamad, N.V.; Ima-Nirwana, S.; Chin, K.Y. Are Oxidative Stress and Inflammation Mediators of Bone Loss Due to Estrogen Deficiency? A Review of Current Evidence. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 1478–1487.
- Kim, O.Y.; Chae, J.S.; Paik, J.K.; Seo, H.S.; Jang, Y.; Cavaillon, J.M.; Lee, J.H. Effects of aging and menopause on serum interleukin-6 levels and peripheral blood mononuclear cell cytokine production in healthy nonobese women. Age 2012, 34, 415–425.
- Srivastava, R.K.; Dar, H.Y.; Mishra, P.K. Immunoporosis: Immunology of Osteoporosis-Role of T Cells. Front. Immunol. 2018, 9, 657.
- Bozec, A.; Zaiss, M.M. T Regulatory Cells in Bone Remodelling. Curr. Osteoporos. Rep. 2017, 15, 121–125.
- Zaiss, M.M.; Sarter, K.; Hess, A.; Engelke, K.; Böhm, C.; Nimmerjahn, F.; Voll, R.; Schett, G.; David, J.P. Increased bone density and resistance to ovariectomy-induced bone loss in FoxP3-transgenic mice based on impaired osteoclast differentiation. Arthritis Rheum 2010, 62, 2328–2338.
- Cline-Smith, A.; Axelbaum, A.; Shashkova, E.; Chakraborty, M.; Sanford, J.; Panesar, P.; Peterson, M.; Cox, L.; Baldan, A.; Veis, D.; et al. Ovariectomy Activates Chronic Low-Grade Inflammation Mediated by Memory T Cells, Which Promotes Osteoporosis in Mice. J. Bone Miner. Res. 2020, 35, 1174–1187.
- Fessler, J.; Husic, R.; Schwetz, V.; Lerchbaum, E.; Aberer, F.; Fasching, P.; Ficjan, A.; Obermayer-Pietsch, B.; Duftner, C.; Graninger, W.; et al. Senescent T-Cells Promote Bone Loss in Rheumatoid Arthritis. Front. Immunol. 2018, 9, 95.
- Pietschmann, P.; Mechtcheriakova, D.; Meshcheryakova, A.; Föger-Samwald, U.; Ellinger, I. Immunology of Osteoporosis: A Mini-Review. Gerontology 2016, 62, 128–137.
- Das, M.; Cronin, O.; Keohane, D.M.; Cormac, E.M.; Nugent, H.; Nugent, M.; Molloy, C.; O’Toole, P.W.; Shanahan, F.; Molloy, M.G.; et al. Gut microbiota alterations associated with reduced bone mineral density in older adults. Rheumatology 2019, 58, 2295–2304.
- Ding, K.; Hua, F.; Ding, W. Gut Microbiome and Osteoporosis. Aging Dis. 2020, 11, 438–447.
- Rios-Arce, N.D.; Schepper, J.D.; Dagenais, A.; Schaefer, L.; Daly-Seiler, C.S.; Gardinier, J.D.; Britton, R.A.; McCabe, L.R.; Parameswaran, N. Post-antibiotic gut dysbiosis-induced trabecular bone loss is dependent on lymphocytes. Bone 2020, 134, 115269.
- Chen, J.; Vitetta, L. The Role of Butyrate in Attenuating Pathobiont-Induced Hyperinflammation. Immune Netw. 2020, 20, e15.
- Wang, J.; Gu, X.; Yang, J.; Wei, Y.; Zhao, Y. Gut Microbiota Dysbiosis and Increased Plasma LPS and TMAO Levels in Patients With Preeclampsia. Front. Cell. Infect. Microbiol. 2019, 9, 409.
- Turner, J.D.; Naylor, A.J.; Buckley, C.; Filer, A.; Tak, P.-P. Fibroblasts and Osteoblasts in Inflammation and Bone Damage. In Stromal Immunology; Owens, B.M.J., Lakins, M.A., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 37–54.
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763.
- Lee, Y.J.; Hong, J.Y.; Kim, S.C.; Joo, J.K.; Na, Y.J.; Lee, K.S. The association between oxidative stress and bone mineral density according to menopausal status of Korean women. Obstet. Gynecol. Sci. 2015, 58, 46–52.
- Wu, J.; Su, J.; Wang, Y.; Chen, J.; Shang, Y.; Li, J. Association between total bilirubin and bone mineral density level in adolescents. BMC Musculoskelet. Disord. 2022, 23, 639.
- Chin, K.Y.; Ima-Nirwana, S. The effects of alpha-tocopherol on bone: A double-edged sword? Nutrients 2014, 6, 1424–1441.
- Chin, K.Y.; Ima-Nirwana, S. Vitamin C and Bone Health: Evidence from Cell, Animal and Human Studies. Curr. Drug Targets 2018, 19, 439–450.
- Maurel, D.B.; Boisseau, N.; Benhamou, C.L.; Jaffre, C. Alcohol and bone: Review of dose effects and mechanisms. Osteoporos. Int. 2012, 23, 1–16.
- Al-Bashaireh, A.M.; Haddad, L.G.; Weaver, M.; Chengguo, X.; Kelly, D.L.; Yoon, S. The Effect of Tobacco Smoking on Bone Mass: An Overview of Pathophysiologic Mechanisms. J. Osteoporos. 2018, 2018, 1206235.
- Agidigbi, T.S.; Kim, C. Reactive Oxygen Species in Osteoclast Differentiation and Possible Pharmaceutical Targets of ROS-Mediated Osteoclast Diseases. Int. J. Mol. Sci. 2019, 20, 3576.
- Domazetovic, V.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Oxidative stress in bone remodeling: Role of antioxidants. Clin. Cases Miner. Bone Metab. 2017, 14, 209–216.
More