The evolution of melanoma, the most aggressive type of skin cancer, is triggered by
driver mutations that are acquired in the coding regions of particularly BRAF (rat fibrosarcoma
serine/threonine kinase, isoform B) or NRAS (neuroblastoma-type ras sarcoma virus) in melanocytes.
Although driver mutations strongly determine tumor progression, additional factors are likely
required and prerequisite for melanoma formation. Melanocytes are formed during vertebrate
development in a well-controlled di erentiation process of multipotent neural crest stem cells
(NCSCs). However, mechanisms determining the properties of melanocytes and melanoma cells
are still not well understood. The nerve growth factor receptor CD271 is likewise expressed in
melanocytes, melanoma cells and NCSCs and programs the maintenance of a stem-like and migratory
phenotype via a comprehensive network of associated genes. Moreover, CD271 regulates phenotype
switching, a process that enables the rapid and reversible conversion of proliferative into invasive or
non-stem-like states into stem-like states by yet largely unknown mechanisms. Here, we summarize
current findings about CD271-associated mechanisms in melanoma cells and illustrate the role of
CD271 for melanoma cell migration and metastasis, phenotype-switching, resistance to therapeutic
interventions, and the maintenance of an NCSC-like state.
- CD271
- melanoma
- neural crest stem cells
- migration
- metastasis
1. Introduction
The acquisition of a metastatic phenotype presents a hallmark in the progression of solid tumors and is rather marked by transcriptomic and epigenetic than genetic changes, mediating the activation or inactivation of molecular programs. The latter determine the progression stages of melanoma and the therapeutic control of tumors. Patients diagnosed with melanoma in non-metastatic stages IA and IIA show a five-year overall survival (OS) of 95.3 ± 0.4% and 78.7 ± 1.2%. The metastasis into regional lymph nodes and the formation of satellite skin metastases are observed in stage IIIA, resulting in a decrease of OS to 69.5 ± 3.7%. The systemic dissemination of melanoma cells throughout the body of stage IV patients leads to metastasis formation in distant organs particularly, lung, liver and brain which is associated with worst outcome and OS to 9.5 ± 1.1% [1]. The adaptation of tumor cells to prevailing microenvironmental conditions that are defined by the composition of growth factors and extracellular matrix (ECM) proteins is prerequisite for the proper establishment of metastases at distant organ sites [2][3]. Several lines of evidence suggest that metastasis is a sequential process [2][4] that likely select for a cellular subset featuring a high metastatic capacity and stem-like phenotype. The latter enables a rapid adaptation to different organ-specific conditions [5]. However, mechanisms driving the metastatic cascade in melanoma are still not well understood, although some promising candidates have been identified.
The nerve growth factor receptor CD271 (p75NTR, NGFR) was identified in 1968, functionally described in 1986, and associated with melanoma aggressiveness and metastasis in the late 1980s and early 1990s. Particularly, Herrmann et al. associated the expression of CD271 with increased invasiveness and brain metastasis of MeWo-70W cells [6][7]. However, CD271 was rediscovered only ~30 years later. In 2010, Boiko et al. established that CD271 labels a subset of melanoma-initiating cells (MICs, aka melanoma stem cells) capable of tumor formation and differentiation. Most strikingly, CD271 showed a mutually exclusive expression with typical melanoma cell surface markers like MART1 and HMB45 [8]. Hence, clinical trials targeting melanoma-specific antigens failed due to the insufficient targeting of MICs, which consequentially promoted tumor relapse. MICs that exhibit expression of CD271 are capable of renewing the tumor mass and give rise to differentiated progeny that feature typical melanocyte/melanoma antigens. Boiko et al. and others provided insight into the role of CD271 in melanoma but also demonstrated that the expression and localization of CD271 like other markers underlie cellular plasticity [8][9]. The expression profiling of melanoma cells with a stable knockdown of CD271 demonstrated for the first time that cellular properties of a subset of melanoma cells is regulated in a CD271-dependent fashion. Subsequent studies have shown that CD271 is part of a network controlling basic properties of melanoma cells.
In the present review, we will summarize recent findings about the role of CD271 in melanoma and demonstrate that CD271 controls basic properties of melanoma cells, particularly migration and metastasis.
2. CD271 in Development
The expression of CD271 is observed at different stages of murine and human development, found earliest in cells of the inner cell mass of the murine blastocyst [10], maintained in murine embryonic stem cells (mESCs) but lost upon differentiation [11]. During vertebrate nervous system development, CD271 labels a large proportion of migrating multipotent neural crest stem cells (NCSCs) capable of self-renewal [12], which arise from the embryonic ectoderm. NCSCs delaminate from the neuroepithelium by epithelial-to-mesenchymal transition (EMT) and migrate along body axes. Migrating neural crest (NC) cells lose the expression of E-cadherin (CDH1) and gain expression of human natural killer-1 (HNK1)/3-beta-glucuronosyltransferase 1 (B3GAT1), CD271, and endothelin receptor type B (EDNRB)[13][14]Figure 1. CD271 in development and melanoma. (a) Simplified schematic representation of neural crest cell delamination and migration and markers of migrating (HNK1/B3GAT1) and non-migrating (CD271, SOX10, FOXD3, CDH1) neural crest stem cells (NCSCs). The regulated differentiation of NCSCs give rise to melanocytes, glia cells, chondrocytes, neurons and smooth muscle cells. (b) Schematic representation of the skin layers (left) and location of CD271 expressing transient-amplifying keratinocytes (CD271+/MITF−/MART1−) and melanocytes (CD271+/MITF+/MART1+, black arrows), right panels. Proliferative cells/melanocytes, aremarked by Ki67 (black arrows). (c) The comparative analysis of different melanoma progression stages (Nevus, primary melanoma; PM, and metastases; MET), normal skin (NS), and melanocytes (MEL) of study GSE46517 revealed increased levels of NGFR/CD271 by trend. (d) The analysis of discordant and concordant metastases revealed the expression of CD271 in metastases of different organs (skin, lymph node; LN and brain; MBM) and the maintenance of CD271 among concordant samples. . The expression of CD271 enables the tracking of NCSCs during early human NC development and the isolation of multiple NCSCs from mammalian fetal peripheral nerve and differentiated human embryonic stem cells (hESCs) [12][13][15]. The hESC-derived NCSCs feature a high expression of CD271, SOX10, PAX3, etc. but a low expression of lineage-specific differentiation markers [15], reviewed in references [12][16]. Likely, the maintenance of multipotent NCSCs during NC development is driven by the transcription factor forkhead box d3 (FOXD3), which acts in concert with CD271 [17][18].
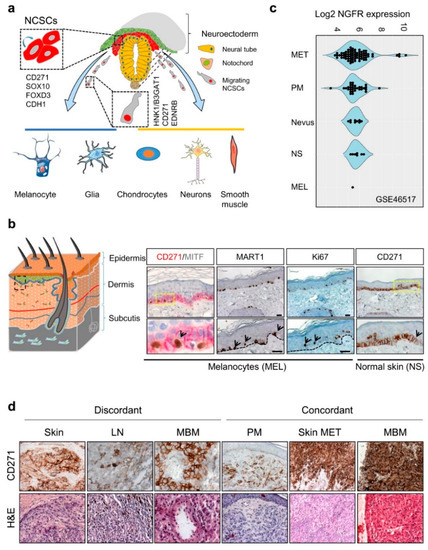
Figure 1. CD271 in development and melanoma. (a) Simplified schematic representation of neural crest cell delamination and migration and markers of migrating (HNK1/B3GAT1) and non-migrating (CD271, SOX10, FOXD3, CDH1) neural crest stem cells (NCSCs). The regulated differentiation of NCSCs give rise to melanocytes, glia cells, chondrocytes, neurons and smooth muscle cells. (b) Schematic representation of the skin layers (left) and location of CD271 expressing transient-amplifying keratinocytes (CD271+/MITF−/MART1−) and melanocytes (CD271+/MITF+/MART1+, black arrows), right panels. Proliferative cells/melanocytes, aremarked by Ki67 (black arrows). (c) The comparative analysis of different melanoma progression stages (Nevus, primary melanoma; PM, and metastases; MET), normal skin (NS), and melanocytes (MEL) of study GSE46517 revealed increased levels of NGFR/CD271 by trend. (d) The analysis of discordant and concordant metastases revealed the expression of CD271 in metastases of different organs (skin, lymph node; LN and brain; MBM) and the maintenance of CD271 among concordant samples.
Following delamination, CD271+/SOX10+/HNK1+/CDH1−/FOXD3− NCSCs migrate through the periphery and give rise to differentiated progeny; among them are muscle cells, glia cells, neurons, and melanocytes [19][20] (Figure 1a). The formation of melanocytes like other NCSC-derivatives is controlled by a network of regulatory transcription factors, particularly SOX10. The genetic ablation of SOX10 and CD271 in a mouse model system consistently led to a complete loss of the melanocyte-differentiation capacity and loss of the NC compartment [21][22]. The EMT that in turn is mediating the delamination of NCSCs from the NC (Figure 1a) is mediated by binding of the transcriptional repressors SNAI1 (Snail), SNAI2 (Slug), and ZEB1 to the E-cadherin/CDH1 promoter, reviewed by Barrallo-Gimeno et al. [23]. First insight into the functional role of CD271 in cell migration was provided by murine models, which enabled the conditional knockout of CD271 in NC cells. The latter revealed a decrease in the sciatic nerve diameter and a deficiency of hematopoiesis, reviewed by Wislet et al. [14]. Although the role of CD271 during melanocyte development and specification , comprehensively reviewed by Douarin et al., White et al. and Blake et al. [19][24][25] is unknown, knockdown studies in melanoma revealed a close relationship of CD271 with FOXD3, SOX2, and SOX10. This may suggest that CD271 promotes the maintenance of a NCSC-state in vivo and at least partially in vitro by the transcriptional control of NCSC-specifier.
3. CD271 in the Skin and Development of Melanoma
CD271 is predominantly expressed in proliferating (Ki67+) and dormant (Ki67−) cells of the stratum basale, particularly melanocytes and transient-amplifying (TA) keratinocyte progenitors [26], (Figure 1b). In the latter, CD271 controls an early and dosage dependent step of keratinocyte differentiation [27]. Albeit the role of CD271 in melanocytes is not completely understood, several lines of evidence suggest that CD271 prevents UV-radiation-induced apoptosis in KIT (c-KIT) and MITF-positive melanocytes via up-regulation of the anti-apoptotic protein BCL-2 . The latter in turn is induced by bioactive NGF that is secreted by keratinocytes, which reside in close proximity to melanocytes. Moreover, levels of NGF and CD271 are increased in keratinocytes and melanocytes in vitro in response to UV-radiation, respectively [28][29][30]. Moreover, levels of CD271 expression are significantly higher in melanoma than in other types of skin cancer like basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) , showing less severe progress. Besides skin melanocytes, a strong expression of CD271 is found in the eye´s choroid and in uveal melanoma .
The formation of melanocytic nevi is the consequence of a hyper-activation of the MAPK pathway, triggered by acquired mutations in BRAF, most prevalently BRAFV600E/K (~15–20% and ~40–50%) [31]. The constitutive activation of BRAF temporally leads to a focal proliferation of melanocytes, which consequently induces a cell cycle arrest and oncogene-induced senescence [32][33][34]. Hence, the loss of tumor suppressor genes, e.g., p16INK4a, PTEN, or TP53, is prerequisite for the transformation of BRAF-mutated melanocytes and formation of melanoma [35][36]. This genetic dependency was recapitulated in a murine model system first established by Dankort et al. In this system, oncogenic BrafV600E was expressed under control of the tyrosinase/Tyr promoter and, hence, was restricted to melanocytes. Whereas the distinct expression of BrafV600E triggered oncogene-induced senescence, the genetic loss of Pten mediated melanoma formation with a high efficacy [36]. Besides Braf, mutated Nras, Kras, or Hras were shown to effectively induce melanoma formation in combination with a genetic ablation of p16Ink4a, p53 and Cdkn2a among other, reviewed by Perez-Guijarro et al. [38]. In addition, activated AKT3 cooperates with BRAFV600E to promote melanocyte transformation, which in turn is associated with a growth factor independent proliferation and enhanced anchorage-independent growth [33]. Although the AKT signaling pathway is frequently activated in melanoma progression, the loss of PTEN predominantly precedes the activation of AKT3 [39].
The identification of CD271 as a marker of MICs by Boiko et al. stimulated the intense investigation of the receptor in the field of melanoma and beyond. Most intriguingly, the expression of CD271 defines a subpopulation of melanoma cells lacking the expression of typical antigens like MART-1, HMB45, melanoma-associated antigens (MAGE) and regulators of proliferation or melanin synthesis e.g., MITF or TYR [8][40]. Likely, the low expression of MITF in CD271+ amelanotic tumors and cell lines is the consequence of a co-expression of CD271 and transcriptional repressors of MITF, e.g., FOXD3 and/or SOX2 [8][17][40]. The investigation of melanoma cell sub fractions retaining the lipophilic dye PKH26 revealed that even the pool of CD271+ cells contains proliferating (CD271+/Ki67+/PKH26-) and slow cycling/label-retaining (CD271+/Ki67−/PKH26+) sub sets [40]. The latter subset showed a higher expression of DNA-repair genes [43] and was linked with resistance to vemurafenib and a brain metastatic phenotype [44][45].
However, the persistence of CD271 expression in tumor cells is unknown and likely indeterminable. In addition, in vitro established tumor-derived melanoma cells exhibit an unstable and fluctuating expression of CD271 which might be stabilized by microenvironmental cues within the tumor and/or are potentially regulated by the circadian clock [46][47]. In addition, the level of CD271 increased in response to stress conditions, e.g., drug-induced DNA-damage [8][9][43][47][48]. Indeed, several melanoma metastases nearly containing ~90–100% of CD271 expressing cells were observed, suggesting the existence of a mechanism that stabilizes or forces CD271 expression in vivo. Consequentially, CD271 regulates key features of melanoma cells; particularly, a stem-like/NCSC-like phenotype and cell migration and plasticity abrogated in melanoma cells with a stable knockdown of CD271 (see Section 5).
However, the role of CD271 in melanoma development and progression is not well defined. Recently, the comparative analysis of gene expression data of CD271+ and CD271− melanoma cells and CD271+ melanocytes identified AKT3 (but not AKT1 or AKT2) among the top up-regulated genes serving as a key mediator of survival [49]. In addition, a pathway analysis revealed SHC1 as interaction partner of CD271 and increased levels of CAMKII in CD271+ cells serving as mediators of activated AKT3 [48]. Consistently, the stable shRNA-mediated knockdown of CD271 decreased the expression of AKT3 [50], suggesting that the concerted action of CD271 and AKT3 may promote the melanocyte transformation and maintenance of early established melanoma cells. However, whether CD271 serves as a crucial supportive factor controlling melanocyte transformation and melanoma progression remains unknown, although levels of CD271 expression increase with progression stages (Figure 1c,d). Only in vivo models will uncover the relevance of CD271 expression in this process. The comparative expression profiling of melanocytes and melanoma cells revealed that CD271 is associated with signaling processes involved in skin development, metabolic hormone processes, cell adhesion and ion channel activity [49]. In addition, the comparison of melanocytes with metastatic melanoma cells revealed a predominant expression of genes associated with interferon response and signaling via E-cadherin (CDH1) in melanocytes and genes involved in metastasis, EMT, and EGFR-signaling in melanoma .
4. Identification of CD271-Associated Genes
Over the past few years, it has been recognized that a subset of melanoma cells features an NCSC-like phenotype [8][40][51]. The stable or transient knockdown of CD271 in melanoma cells revealed that the NCSC-like phenotype is maintained by expression of CD271 (Figure 1d) which in turn controls the levels of downstream targets, e.g., SOX10, FOXD3, and SOX2 [12][40][52]. The loss of CD271 consequentially induced a severe impairment of cell survival and proliferation, most likely caused by a loss of genomic integrity that in turn was associated with a reduced expression of DNA-repair genes [40][43] and revealed a mutually exclusive expression with inhibitors of metastasis, KISS1, and DMBT1 (Figure 2) [43]. In addition, the decrease of CD271 significantly reduced the capability of tumor formation in vivo and cell migration in vitro [40][44][50]. On the other hand, the overexpression of CD271 increased levels of SOX2 and RHOJ and the propensity of migration and metastasis of A375 cells [43][53]; however, the underlying mechanisms are poorly understood. These experiments strongly suggest a more comprehensive role of CD271, serving as determinant of melanoma cell properties independent from the presence of a BRAFV600E-mutation. The presence of the latter was not sufficient to prevent melanoma cells from apoptosis induced by the loss of CD271 expression [44]. Melanoma cell properties are likely controlled by a concerted cooperation of CD271 and SOX10 as the down regulation of either CD271 or SOX10 showed comparable effects [22]. Hence, CD271 and SOX10 share a common set of associated genes or targets, e.g., MITF, CDKN1A, TP53, and CCNA1 [40]. Interestingly, the loss of SOX10 reduced the level of CD271, suggesting a mutual dependency. Concordantly, the genetic ablation in Tyr::NrasQ61K mice demonstrated that Sox10 is required for melanoma formation and proliferation, survival, and tumor maintenance [22]. However, if the genetic ablation of CD271 in melanocytes impairs melanoma formation has not yet been investigated.
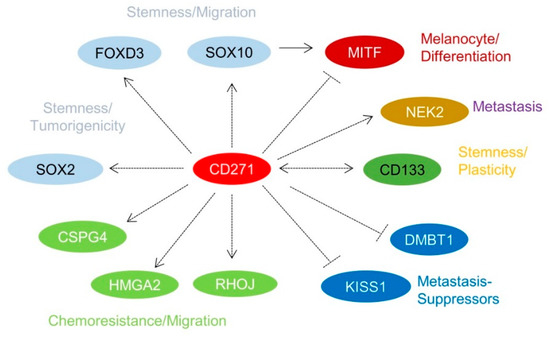
5. CD271 in Migration, Metastasis, and Cellular Plasticity
The systemic dissemination of tumor cells throughout the body presents a hallmark of tumor progression and is still the leading cause of cancer-related death. Particularly the sequential formation of metastases at distant organs is associated with the loss of disease control and poor prognosis, as reviewed by Ackermann et al. [54]. Brain metastases (BM) are most challenging [55] and develop in 20–40% of melanoma cases during the course of disease, with unclear subtype specificity, as reviewed by Redmer [56]. The efficient systemic dissemination of melanoma cells relies on the maintenance of a migratory phenotype, which is likely inherited from NC cells [57][58]. Likely, the metastatic niche plays an important role for the maintenance of the migratory phenotype. In 1995, Marchetti et al. demonstrated that the ligands for neurotrophin receptors CD271 and tropomyosin-related kinases (TRKs, see Section 6), nerve growth factor (NGF), and neurotrophin 3 (NTF3) were expressed by niche cells within the brain but not brain metastatic melanoma cells [59]. On the contrary, the latter expressed CD271 and TRKC and featured migration at the invasion front likely in a paracrine response. The relevance of this dependency was demonstrated by Truzzi et al., who observed increased migration of metastatic melanoma cells after stimulation with NGF, NTF3, and NTF4 [60]. Although the in vitro situation might not directly reflect mechanisms of tumor cell migration within the brain, these studies suggest a role of neurotrophins in cell migration. More than a decade later, studies by Boiko et al. evidenced that the expression of CD271 mediates the stemness of melanoma cells and serves as a regulator of metastasis [8]. Later functional studies established that CD271 acts as a key regulator of the NCSC-like state in melanoma that, in turn, programs the migratory phenotype [40][50]. The expression profiling and gene-set enrichment analysis (GSEA) of melanoma cells with forced expression of CD271 revealed the association with genes conferring metastasis and relapse. Concordantly, live-cell imaging-based scratch-wound assays revealed that melanoma cells with a high endogenous level or forced expression of CD271 featured a significantly enhanced migration into the scratch wound than cells with a low expression and, consistently, the knockdown of CD271 decreased the migratory capacity [43][50]. Among the genes predicting either melanoma metastasis or relapse were hyaluronan mediated motility receptor (HMMR), NIMA related kinase 2 (NEK2), and DNA-repair genes. HMMR and NEK2 were associated with increased migration, metastasis, and poor survival in cancer [43][61][62][63][64][65][66][67][68], and Kauffmann et al. associated metastasis of melanoma with a high expression of DNA-repair genes and pathways [69]. We observed increased levels of DNA-repair genes RAD21, MSH6, and RAD51 in CD271+ but not CD271− MACSed cells hence, the DNA-repair capacity is potentially regulated in a CD271-dependent manner [43]. Mutant BRAFV600 serves as yet another driver of metastasis as mutant melanoma cells featured a higher endogenous migration capacity in vitro than wild type (wt) cells [43][70]. Although the level of CD271 in tumors is likely unstable [47], several processes raising the expression of CD271 have been identified in vitro. Particularly, cellular stress induced by therapeutic interventions [43][44][71], hypoxia [72][73], or inflammation [74][75][76][77] increased the expression of CD271. Chemotherapeutics have been used for more than 40 years in the first-line treatment of melanoma and are still in clinical use, particularly applied to patients lacking druggable mutations or patients who became refractory to immune checkpoint inhibitors [78]. However, DNA-damage inducing chemotherapeutic drugs increased the cell surface and total levels of CD271 after 24 hours of treatment in drug-sensitive (parental, Par) MeWo cells . The significant increase in the DNA-damage and p53-pathways likely suggest a role of both regulating the levels of CD271 in vitro and in vivo. Besides mediators of the DNA-damage response, NGF and BDNF were up regulated in a short-term (24h) response to cisplatin, potentially triggering autocrine signaling processes e.g., migration. In addition, increased expression of CD271 and NGF was observed in chemo-resistant MeWo cells featuring increased migration [43]. Concordantly, the selective inhibitor of oncogenic BRAFV600E/K vemurafenib mediated resistance via increased expression of CD271 [44].
In summary, therapeutic interventions select for highly migratory and metastatic melanoma cells, likely via the modification of levels of CD271. This statement is underpinned by the intriguing finding that the injection of a neutralizing antibody raised against CD271 blocks melanoma metastasis in a mouse model system [79].
Recently, the expression of CD271 was recognized as a regulator of phenotype switching, a process that enables the rapid and reversible conversion of non-stem-like into stem-like or proliferative into invasive states. The latter was associated with the AXLhigh/MITFlow program [53][54][55][56][57][58]. In addition, Hoek et al. specified the proliferative/low-migratory phenotype by (1) the levels of MITF-targets (TYR, DCT, PMEL, MLANA), (2) NC-related genes (SOX10, TFAP1A, EDNRB), and (3) the sensitivity toward the growth-inhibitory effect of TGF-β and the proportion of Ki67-positive cells [80][81]. Contrary, the migratory/low-proliferative phenotype was specified by EMT-inducers ZEB1, TWIST1, and c-JUN as well as CD271 and AXL [53][82]. The conditional overexpression of CD271 in melanoma cell lines impaired the proliferation in vitro and in vivo but enhanced metastasis formation at distant organs. Furthermore, Restivo et al. demonstrated that the manipulation of levels of CD271 was sufficient to control the switch between proliferation (“growing”) and invasion (“going”) of melanoma cells. Furthermore, Restivo et al. suggested that the interaction of CD271 with TRKA and the release of the CD271 intracellular domain (ICD) serve as responsible mechanisms, leading to the loss of adhesion via up regulation of cholesterol production and impaired proliferation [53]. The mechanism regulating the switch of CD271high into MITFhigh cells and vice versa is likely found in all organ-metastases and proceeds via a transient CD271+/MITF+ cell state. Consistently, the analysis of melanoma BM (MBM) revealed that CD271high cells exhibit a lack of expression of MITF and MITF-targets [50] but feature a high enrichment of CD271-target/associated genes (study GSE50493 [83]) and genes associated with an NCSC-like phenotype and cell migration .
Several mechanisms, which trigger and control cellular plasticity and phenotype switching, are plausible, besides CD271-ICD-regulated processes. Tumor hypoxia is induced by low oxygen levels triggering not only the formation of new blood vessels but also metastasis, as reviewed by Rankin et al. [84]. Hypoxia-induced metastasis is a complex process driven by several mediators. Widmer et al. demonstrated that the modulation of the hypoxia-inducible factor 1α (HIF1α) was sufficient to induce the dedifferentiation and phenotype switching of proliferative into invasive melanoma cells, referring to the AXL/MITF axis [85]. The stabilization of HIF1α under hypoxic conditions favored the invasive AXLhigh state and the absence or low levels of HIF1α promoted the proliferative MITFhigh phenotype.
Changes in the growth factor environment yet present another mechanism, triggering the rapid interconversion of stem-like states, marked by CD271, CD133 [86] or CD271/CD133 [40]. We observed that cells with a double positive (CD271+/CD133+) as well as a double negative (CD271−/CD133−) phenotype were capable of re-establishing the cellular heterogeneity of the initial cell culture within three days. Moreover, a phenotype switch of CD271+/CD133− to CD271+/CD133+ cells was induced in the absence of serum and FGF2, which was prevented in cells with a stable knockdown of CD271. Whether CD133+ stem-like cells indeed directly evolved from CD271+ cells or presented a differentiated progeny of yet another stem-like state is unknown. The shRNA-mediated down regulation of CD133 or CD271 in melanoma cells revealed that both markers are inversely correlated, suggesting that CD133 suppresses genes associated with CD271, e.g., AXL and FOXD3 and vice versa [40][86].
Although methods enabling the tracing of melanoma cell phenotypes in vitro exist, the monitoring of cues inducing phenotype switching within tumors, particularly in response to certain in vivo conditions, e.g., therapeutic interventions or hypoxia, remains challenging. Reporter systems may enable the in vivo tracing of phenotype switching of stem-like cells and their participation in tumor formation and metastasis. The combination of gene-specific 3′-UTR sequences fused to GFP provide insight into mechanisms maintaining stem-like cells (Figure 3a), e.g., regulated by microRNA binding. The FACS-enrichment (Figure 3b) of melanoma cells with a stable genomic integration of a 3´-UTR-CD271-GFP reporter yields in a pure GFP+ subset (Figure 3c, upper panels). However, only a minority of GFP+ cells is maintained upon culturing in two dimensional (2D) (Figure 3c, lower panels) or three dimensional (3D) systems (Figure 3d). Hence, the content of putative CD271+ stem-like cells within cell culture systems is tightly controlled by mechanisms balancing differentiation and dedifferentiation.
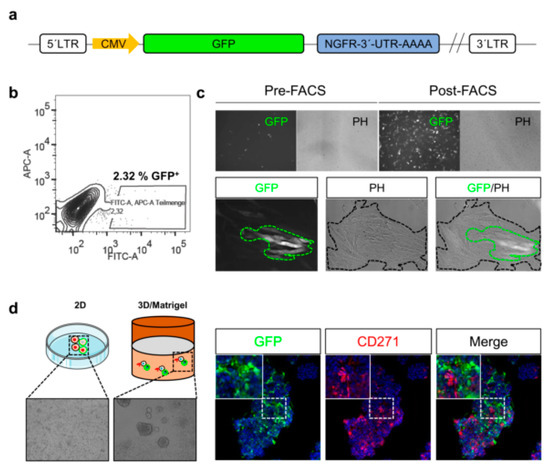
In summary, these data suggest that CD271 likely controls melanoma cell migration and metastasis via phenotype switching which includes the suppression of MITF-targets. The comparison of MITF expression levels reveal that metastases and primary tumors featuring proliferative and invasive cell states with no organ-specific prevalence . Hence, the expression of MITF/targets is suppressed during metastasis and rises during the formation of secondary tumors. In contrast, the expression of CD271 increases to facilitate metastasis, invasiveness and decreases once the disseminated cells arrived at distant organ sites. Moreover, a subset of metastases may permanently exist in a CD271high/MITFlow state featuring a high aggressiveness and metastatic capacity. The persistence of the CD271high/MITFlow state in vivo is unknown; however, therapeutic interventions potentially influencing the equilibrium of invasiveness and proliferation might select for the most favorable state and consequentially foster specific metastatic routes, e.g., metastasis to the brain [87].
6. Signaling Mechanisms of CD271 in the Central Nervous System
CD271 is widely expressed in different cell types of the central nervous system (CNS), e.g., neurons and schwann cells, regulating a plethora of processes like survival and apoptosis, myelination and axonal regeneration in response to specific ligands like the neurotrophins (NGF, BDNF, NTF3, and NTF4). In addition, CD271 is expressed in several cellular compartments, is localized in the nucleus following neurotrophin-induced cleavage, the nuclear pore and shows membrane localization [76][88][89][90].
In the subventricular zone, the place of adult neurogenesis, CD271 labels a subset of progenitor cells, which are maintained in response to BDNF [90][91][92]. Consistently, the expression of CD271 is down regulated upon neuronal differentiation of progenitor cells in a rodent model system [92]. In addition, astrocytes gain expression of CD271 in response to injury and neuroinflammation that in turn is triggered in response to injury, seizure, toxic metabolites and tumor cells, as reviewed by Meeker et al. [93]. The activation of astrocytes, known as astrogliosis strongly determines the maintenance of tumor cells within the brain as reactive astrocytes secrete chemokines, cytokines and growth factors supporting tumor cell growth and migration, comprehensively reviewed by Sofroniew et al. [94]. The exact role of CD271 in astrogliosis is not well understood, although it might affect astrocyte proliferation [75]. CD271 lacks in a kinase-domain and regulates signaling processes in conjunction with co-receptors and/or the recruitment of signaling mediators that bind to the death domain of CD271 homodimers (Figure 4), as reviewed by Meeker et al. [93].
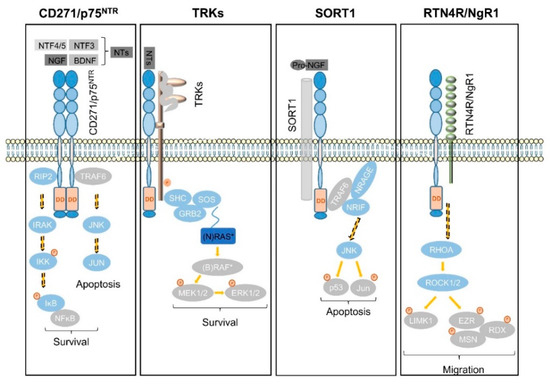
Signaling via CD271 (p75NTR) homodimers mediates the activation of NFĸB signaling, driving the expression of pro-survival genes via the recruitment of receptor-interacting serine/threonine-protein kinase 2 (RIP2). In the absence of the latter that competes with TRAF6 for binding to CD271, neurotrophins trigger the activation of c-jun-n-terminal kinase (JNK) to drive apoptosis in neurons [95] (Figure 4, first panel). In addition, the association of CD271 with tropomyosin-related kinases (TRKs) results in the generation of high-affinity neurotrophin binding sites and mediates cell survival in a mitogen-activated kinase (MAPK)-dependent manner via adaptor proteins SHC, GRB2, and SOS (Figure 4, second panel). Besides MAPK signaling, the CD271/TRK heterodimers signal trough PI3K and PLCγ [14]. Sortilin (SORT1), belongs to the vacuolar protein sorting 10 family (Vps10) and is mainly found within late endosomes derived from the trans-golgi network, controlling the anterograde trafficking of neurotrophin receptors and secretion of pro-BDNF [96]. The binding of neurotrophin precursors like pro-NGF or pro-BDNF to the CD271/SORT1 complex triggers apoptosis via adaptor proteins TRAF6, NRAGE and NRIF in neurons [97] (Figure 4, third panel), reviewed in references [16][98]. Moreover, CD271 forms a complex with Nogo receptor (RTN4R), MAG, and LINGO1 (not shown), inducing the release of RhoA from Rho GDP-dissociation inhibitor (RhoGDI) and subsequently activates the RhoA/ROCK signaling pathway. The latter in turn regulates the dynamic of the actin cytoskeleton (reviewed by Fujita et al. [99]), determining cell shape and motility via phosphorylation and activation of the Ser/Thr kinase LIM. Activated LIM in turn phosphorylates and inactivates the actin de-polymerization factor cofilin and mediates the inhibition of neurite outgrowth, reviewed in [93][99]. Furthermore, LIM activates the ERM proteins radixin (RDX), ezrin (EZR), and moesin (MSN) (Figure 4, fourth panel), which are important regulators of microvilli formation, cell adhesion, and membrane ruffling (reviewed by Tsukita et al.[100]) and potential drivers of cancer progression (reviewed by Clucas et al. [101]).
However, CD271 signaling mechanisms show high context dependence, hence facilitate cellular properties in relation to the abundance of co-receptors, ligands, and recruited adaptor proteins that mediate signaling. Yet-identified signaling mechanisms in the CNS are more comprehensively discussed by Roux and Barker [102].
7. Signaling of CD271 in Melanoma and Other Cancer Types
During central nervous system (CNS) development, the NC gives rise to several differentiated progeny, besides melanocytes like glia cells, which in turn give rise to astrocytes. Several lines of evidence suggest that particular cancer entities have adopted molecular features used by NC cells (reviewed by Maguire et al. [103].
The first evidence that CD271 consequentially determines melanoma cell behavior was provided by Shonukan et al. who demonstrated the specific interaction of CD271 with the actin cytoskeleton via the actin-bundling protein fascin (FSCN1). This interaction facilitates the migration of melanoma cells in a NGF and pro-NGF dependent manner [104]. Moreover, Marchetti et al. observed the activation of heparanase (HPSE1) expression in melanoma triggered by NGF and CD271 in a dose-dependent manner and even in the absence of TRKA[6][105][106]. HPSE1 promoted melanoma cell invasion and in turn was reported to induce PI3K/AKT signaling and probably promote tumor cell migration modulated by PLEKHA5, which specifically interacts with phosphoinositides [107]. Furthermore, Restivo et al. suggested that the binding of NGF to the complex of CD271 and TRKA triggered cell detachment and cleavage of CD271, yielding in the CD271-ICD that, in turn, induced the “growing” to “going” switch [53]. Moreover, the cleavage of CD271 by ADAM10 or ADAM17 controls the formation of the intracellular domain fragment (ICD) or carboxyterminal fragment (CTF), respectively. Both fragments mediate[108][109] resistance to apoptosis mediated by TRAIL (CTF) or triggered by anoikis (ICD) in breast cancer and/or HNSCC (Figure 5b). The malignant transformation of glia cells causes the development of astrocytoma including glioblastoma, the major forms of primary brain tumors in adulthood. Although astrocytes lack expression of CD271, several processes within the CNS like astrogliosis increase the level of CD271 on astrocytes. Like for melanoma, the contribution of CD271 to the malignant transformation of astrocytes is completely unknown. Subsets of glioblastoma and astrocytoma (Figure 5a, not shown) as well as primary tumors and BM of melanoma (Figure 5a, left panels, and Figure 1d) and lung squamous cell carcinoma (LSCC) (Figure 5a, right panels and ) feature a high expression of CD271 [110][111], as reviewed by Wrensch et al. [112]. Concordantly, high levels of CD271 drive migration and invasion of glioblastoma cells, mediated via the PDZ and LIM-domain protein PDLIM1 [111][113], (Figure 5b). The phosphorylation of CD271 at S303/S304 by protein kinase a (PKA) in turn promoted glioblastoma invasiveness and the translocation of CD271 to lipid rafts in neurons and potentially in glioblastoma [113][114].
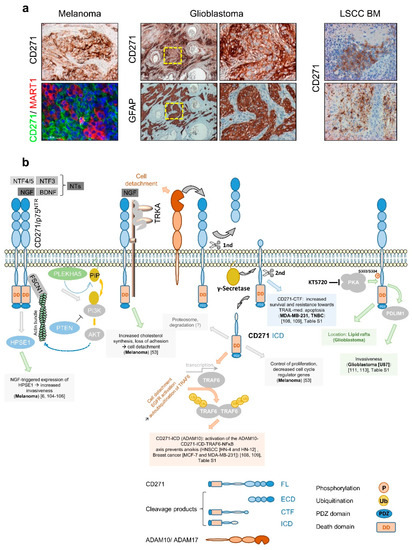
In addition, the expression of CD271 was associated with increased migration/invasion and metastasis of head and neck cancer, hypopharyngeal cancer, and oral squamous cell carcinoma [115][116][117] and was associated with poor prognosis of LSCC [118] and urothelial carcinoma (TCGA, human protein atlas. Strikingly, the tumor formation of engrafted hypopharyngeal cancer and melanoma cells was blocked by a humanized anti-CD271 antibody [119]. In contrast, a high expression of CD271 in medulloblastoma, esophageal squamous cell carcinoma, gastric cancer , and prostate cancer [120] was associated with a favorable tumor state.
In summary, CD271 defines properties of tumor cells of several cancer entities, often associated with a poor outcome. Additional studies are needed to uncover molecular mechanisms that are common and distinct to specific cancer entities.
8. Therapeutic Targeting of CD271
Signaling mechanisms of CD271 in melanoma and other tumor entities are still not well understood, making the specific targeting challenging. As CD271 regulates survival and apoptosis of neurons in conjunction with co-receptors and in dependence of the availability of ligands within the CNS, the direct targeting of either CD271 or neurotrophins seems impossible. Nevertheless, commercially available small molecule drugs like non-peptide inhibitors preventing NGF from binding to CD271; PD90780, Ro 08-2750, and peptide inhibitor NTR368 have not yet been tested in vivo. However, a non-peptide ligand of CD271, LM11A-31 was administered to patients suffering from mild to moderate Alzheimer’s disease in a clinical trial (NCT03069014). The inhibitor potentially inhibits CD271-mediated apoptosis of neurons.
Yet, the most intriguing finding by Ngo et al. suggests that the blocking of CD271 by neutralizing antibodies prevents melanoma metastasis in vivo [79]. Recently, Morita et al. reported that a humanized anti-CD271 antibody was sufficiently blocking the tumor growth of MeWo graft models [119]. However, the systemic application of CD271 neutralizing antibodies like inhibitors might negatively interfere with processes within the CNS. The targeting of CD271 is a promising route potentially preventing melanoma from metastasis. Two clinical pilot studies investigating whether T cells genetically engineered to express NGFR/CD271 improve the recognition and eradication of melanoma cells in patients with stage III–IV melanoma, started in 2012 and 2013 (NCT01955460, NCT01740557). It is likely that the adoptive transfer of autologous tumor infiltrating lymphocytes (TIL) is superior to other therapeutic interventions due to higher selectivity [121].
9. Conclusions and Remarks
The nerve growth factor receptor CD271 regulates the survival and apoptosis of neurons within the CNS and basic properties of melanoma cells like tumorigenicity, plasticity, and self-renewal. Moreover, the molecular programs determining the migratory capacity of NCSCs likely drive the migration and metastasis of melanoma cells. The expression of CD271 increases in response to therapeutic drugs, inflammation, and stress and mediates drug resistance. Hence, CD271 likely acts as a key mediator of therapy-induced invasion and metastasis in melanoma and other cancer types. Although the molecular programs controlling the survival, apoptosis, and migration of cells in the CNS are well understood, the signaling mechanisms of CD271 in cancer remain elusive due to the high diversity of modes of signaling. The latter is strongly determined by the levels of co-receptors, ligands adaptor proteins that are recruited to the cytoplasmic domain of CD271. CD271-expressing brain-metastatic cells exhibit a more aggressive phenotype and likely benefit from close proximity to reactive astrocytes. All these findings strongly motivate the therapeutic targeting; however, the understanding of the signaling mechanisms that are directly controlled by CD271 is a prerequisite for the development of therapeutic drugs.
References
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199. [Google Scholar] [CrossRef] [PubMed]
- Obenauf, A.C.; Massague, J. Surviving at a distance: Organ-specific metastasis. Trends Cancer 2015, 1, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. Metastatic colonization: Settlement, adaptation and propagation of tumor cells in a foreign tissue environment. Semin. Cancer Biol. 2011, 21, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.L.; Menter, D.G.; Hamada, J.; Marchetti, D.; Nakajima, M.; Nicolson, G.L. Mediation of ngf-stimulated extracellular matrix invasion by the human melanoma low-affinity p75 neurotrophin receptor: Melanoma p75 functions independently of trka. Mol. Biol. Cell 1993, 4, 1205–1216. [Google Scholar] [CrossRef]
- Marchetti, D.; Menter, D.; Jin, L.; Nakajima, M.; Nicolson, G.L. Nerve growth factor effects on human and mouse melanoma cell invasion and heparanase production. Int. J. Cancer 1993, 55, 692–699. [Google Scholar] [CrossRef]
- Boiko, A.D.; Razorenova, O.V.; van de Rijn, M.; Swetter, S.M.; Johnson, D.L.; Ly, D.P.; Butler, P.D.; Yang, G.P.; Joshua, B.; Kaplan, M.J.; et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor cd271. Nature 2010, 466, 133–137. [Google Scholar] [CrossRef]
- Quintana, E.; Shackleton, M.; Foster, H.R.; Fullen, D.R.; Sabel, M.S.; Johnson, T.M.; Morrison, S.J. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 2010, 18, 510–523. [Google Scholar] [CrossRef]
- Vandamme, N.; Berx, G. From neural crest cells to melanocytes: Cellular plasticity during development and beyond. Cell Mol. Life Sci. 2019, 76, 1919–1934. [Google Scholar] [CrossRef]
- Moscatelli, I.; Pierantozzi, E.; Camaioni, A.; Siracusa, G.; Campagnolo, L. P75 neurotrophin receptor is involved in proliferation of undifferentiated mouse embryonic stem cells. Exp. Cell Res. 2009, 315, 3220–3232. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; White, P.M.; Zock, C.; Anderson, D.J. Prospective identification, isolation by flow cytometry, and in vivo self-renewal of multipotent mammalian neural crest stem cells. Cell 1999, 96, 737–749. [Google Scholar] [CrossRef]
- Betters, E.; Liu, Y.; Kjaeldgaard, A.; Sundstrom, E.; Garcia-Castro, M.I. Analysis of early human neural crest development. Dev. Biol. 2010, 344, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Wislet, S.; Vandervelden, G.; Rogister, B. From neural crest development to cancer and vice versa: How p75(ntr) and (pro)neurotrophins could act on cell migration and invasion? Front. Mol. Neurosci. 2018, 11, 244. [Google Scholar] [CrossRef]
- Lee, G.; Kim, H.; Elkabetz, Y.; al Shamy, G.; Panagiotakos, G.; Barberi, T.; Tabar, V.; Studer, L. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat. Biotechnol. 2007, 25, 1468–1475. [Google Scholar] [CrossRef]
- Tomellini, E.; Lagadec, C.; Polakowska, R.; le Bourhis, X. Role of p75 neurotrophin receptor in stem cell biology: More than just a marker. Cell Mol. Life Sci. 2014, 71, 2467–2481. [Google Scholar] [CrossRef]
- Mundell, A.N.; Labosky, P.A. Neural crest stem cell multipotency requires foxd3 to maintain neural potential and repress mesenchymal fates. Development 2011, 138, 641–652. [Google Scholar] [CrossRef]
- Teng, L.; Mundell, N.A.; Frist, A.Y.; Wang, Q.; Labosky, P.A. Requirement for foxd3 in the maintenance of neural crest progenitors. Development 2008, 135, 1615–1624. [Google Scholar] [CrossRef]
- Douarin, L.M.N.; Dupin, E. Multipotentiality of the neural crest. Curr. Opin. Genet. Dev. 2003, 13, 529–536. [Google Scholar] [CrossRef]
- Mort, R.L.; Jackson, I.J.; Patton, E.E. The melanocyte lineage in development and disease. Development 2015, 142, 620–632. [Google Scholar] [CrossRef]
- Britsch, S.; Goerich, D.E.; Riethmacher, D.; Peirano, R.I.; Rossner, M.; Nave, K.A.; Birchmeier, C.; Wegner, M. The transcription factor sox10 is a key regulator of peripheral glial development. Genes Dev. 2001, 15, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Shakhova, O.; Zingg, D.; Schaefer, S.M.; Hari, L.; Civenni, G.; Blunschi, J.; Claudinot, S.; Okoniewski, M.; Beermann, F.; Mihic-Probst, D.; et al. Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat. Cell Biol. 2012, 14, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Barrallo-Gimeno, A.; Nieto, M.A. The snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [PubMed]
- White, M.R.; Zon, L.I. Melanocytes in development, regeneration, and cancer. Cell Stem Cell 2008, 3, 242–252. [Google Scholar] [CrossRef]
- Blake, J.A.; Ziman, M.R. Pax genes: Regulators of lineage specification and progenitor cell maintenance. Development 2014, 141, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Pincelli, C. P75 neurotrophin receptor in the skin: Beyond its neurotrophic function. Front. Med. (Lausanne) 2017, 4, 22. [Google Scholar] [CrossRef]
- Truzzi, F.; Saltari, A.; Palazzo, E.; Lotti, R.; Petrachi, T.; Dallaglio, K.; Gemelli, C.; Grisendi, G.; Dominici, M.; Pincelli, C.; et al. Cd271 mediates stem cells to early progeny transition in human epidermis. J. Investig. Dermatol. 2015, 135, 786–795. [Google Scholar] [CrossRef]
- Peacocke, M.; Yaar, M.; Mansur, C.P.; Chao, M.V.; Gilchrest, B.A. Induction of nerve growth factor receptors on cultured human melanocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 5282–5286. [Google Scholar] [CrossRef]
- Yaar, M.; Grossman, K.; Eller, M.; Gilchrest, B.A. Evidence for nerve growth factor-mediated paracrine effects in human epidermis. J. Cell Biol. 1991, 115, 821–828. [Google Scholar] [CrossRef]
- Tron, V.A.; Coughlin, M.D.; Jang, D.E.; Stanisz, J.; Sauder, D.N. Expression and modulation of nerve growth factor in murine keratinocytes (pam 212). J. Clin. Investig. 1990, 85, 1085–1089. [Google Scholar] [CrossRef]
- Menzies, A.M.; Haydu, L.E.; Visintin, L.; Carlino, M.S.; Howle, J.R.; Thompson, J.F.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Distinguishing clinicopathologic features of patients with v600e and v600k braf-mutant metastatic melanoma. Clin. Cancer Res. 2012, 18, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of braf mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.; Sharma, A.; Madhunapantula, S.V.; Robertson, G.P. Akt3 and mutant v600e b-raf cooperate to promote early melanoma development. Cancer Res. 2008, 68, 3429–3439. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the braf gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Shain, A.H.; Bastian, B.C. Author correction: From melanocytes to melanomas. Nat. Rev. Cancer 2020, 20, 355. [Google Scholar] [CrossRef]
- Dankort, D.; Curley, D.P.; Nelson, A.N.; Karnezis, W.E.; Damsky, J.; You, R.A.D.; Bosenberg, M. Braf(v600e) cooperates with pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef]
- Perez-Guijarro, E.; Day, C.P.; Merlino, G.; Zaidi, M.R. Genetically engineered mouse models of melanoma. Cancer 2017, 123, 2089–2103. [Google Scholar] [CrossRef]
- Stahl, J.M.; Sharma, A.; Cheung, M.; Zimmerman, M.; Cheng, J.Q.; Bosenberg, M.W.; Kester, M.; Sandirasegarane, L.; Robertson, G.P. Deregulated akt3 activity promotes development of malignant melanoma. Cancer Res. 2004, 64, 7002–7010. [Google Scholar] [CrossRef]
- Redmer, T.; Welte, Y.; Behrens, D.; Fichtner, I.; Przybilla, D.; Wruck, W.; Yaspo, M.L.; Lehrach, H.; Schafer, R.; Regenbrecht, C.R. The nerve growth factor receptor cd271 is crucial to maintain tumorigenicity and stem-like properties of melanoma cells. PLoS ONE 2014, 9, e92596. [Google Scholar] [CrossRef]
- Adameyko, I.; Lallemend, F.; Furlan, A.; Zinin, N.; Aranda, S.; Kitambi, S.S.; Blanchart, A.; Favaro, R.; Nicolis, S.; Lubke, M.; et al. Sox2 and mitf cross-regulatory interactions consolidate progenitor and melanocyte lineages in the cranial neural crest. Development 2012, 139, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M. Mitf in melanoma: Mechanisms behind its expression and activity. Cell Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Redmer, T.; Walz, I.; Klinger, B.; Khouja, S.; Welte, Y.; Schafer, R.; Regenbrecht, C. The role of the cancer stem cell marker cd271 in DNA damage response and drug resistance of melanoma cells. Oncogenesis 2017, 6, e291. [Google Scholar] [CrossRef] [PubMed]
- Lehraiki, A.; Cerezo, M.; Rouaud, F.; Abbe, P.; Allegra, M.; Kluza, J.; Marchetti, P.; Imbert, V.; Cheli, Y.; Bertolotto, C.; et al. Increased cd271 expression by the nf-kb pathway promotes melanoma cell survival and drives acquired resistance to braf inhibitor vemurafenib. Cell Discov. 2015, 1, 15030. [Google Scholar] [CrossRef] [PubMed]
- Zubrilov, I.; Sagi-Assif, O.; Izraely, S.; Meshel, T.; Ben-Menahem, S.; Ginat, R.; Pasmanik-Chor, M.; Nahmias, C.; Couraud, P.O.; Hoon, D.S.; et al. Vemurafenib resistance selects for highly malignant brain and lung-metastasizing melanoma cells. Cancer Lett. 2015, 361, 86–96. [Google Scholar] [CrossRef]
- Baeza-Raja, B.; Eckel-Mahan, K.; Zhang, L.; Vagena, E.; Tsigelny, I.F.; Sassone-Corsi, P.; Ptacek, L.J.; Akassoglou, K. P75 neurotrophin receptor is a clock gene that regulates oscillatory components of circadian and metabolic networks. J. Neurosci. 2013, 33, 10221–10234. [Google Scholar] [CrossRef]
- Boyle, S.E.; Fedele, C.G.; Corbin, V.; Wybacz, E.; Szeto, P.; Lewin, J.; Young, R.J.; Wong, A.; Fuller, R.; Spillane, J.; et al. Cd271 expression on patient melanoma cells is unstable and unlinked to tumorigenicity. Cancer Res. 2016, 76, 3965–3977. [Google Scholar] [CrossRef]
- Cheli, Y.; Bonnazi, V.F.; Jacquel, A.; Allegra, M.; de Donatis, G.M.; Bahadoran, P.; Bertolotto, C.; Ballotti, R. Cd271 is an imperfect marker for melanoma initiating cells. Oncotarget 2014, 5, 5272–5283. [Google Scholar] [CrossRef]
- Filipp, F.V.; Li, C.; Boiko, A.D. Cd271 is a molecular switch with divergent roles in melanoma and melanocyte development. Sci. Rep. 2019, 9, 7696. [Google Scholar] [CrossRef]
- Radke, J.; Rossner, F.; Redmer, T. Cd271 determines migratory properties of melanoma cells. Sci. Rep. 2017, 7, 9834. [Google Scholar] [CrossRef]
- Civenni, G.; Walter, A.; Kobert, N.; Mihic-Probst, D.; Zipser, M.; Belloni, B.; Seifert, B.; Moch, H.; Dummer, R.; van den Broek, M.; et al. Human cd271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011, 71, 3098–3109. [Google Scholar] [CrossRef] [PubMed]
- Mandalos, N.; Rhinn, M.; Granchi, Z.; Karampelas, I.; Mitsiadis, T.; Economides, A.N.; Dolle, P.; Remboutsika, E. Sox2 acts as a rheostat of epithelial to mesenchymal transition during neural crest development. Front. Physiol. 2014, 5, 345. [Google Scholar] [CrossRef] [PubMed]
- Restivo, G.; Diener, J.; Cheng, P.F.; Kiowski, G.; Bonalli, M.; Biedermann, T.; Reichmann, E.; Levesque, M.P.; Dummer, R.; Sommer, L. Publisher correction: The low affinity neurotrophin receptor cd271 regulates phenotype switching in melanoma. Nat. Commun. 2018, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, J.; Frutschi, M.; Kaloulis, K.; McKee, T.; Trumpp, A.; Beermann, F. Metastasizing melanoma formation caused by expression of activated n-rasq61k on an ink4a-deficient background. Cancer Res. 2005, 65, 4005–4011. [Google Scholar] [CrossRef] [PubMed]
- Sandru, A.; Voinea, S.; Panaitescu, E.; Blidaru, A. Survival rates of patients with metastatic malignant melanoma. J. Med. Life 2014, 7, 572–576. [Google Scholar]
- Redmer, T. Deciphering mechanisms of brain metastasis in melanoma-the gist of the matter. Mol. Cancer 2018, 17, 106. [Google Scholar] [CrossRef]
- Bailey, C.M.; Morrison, J.A.; Kulesa, P.M. Melanoma revives an embryonic migration program to promote plasticity and invasion. Pigment Cell Melanoma Res. 2012, 25, 573–583. [Google Scholar] [CrossRef]
- Larribere, L.; Utikal, J. Stem cell-derived models of neural crest are essential to understand melanoma progression and therapy resistance. Front. Mol. Neurosci. 2019, 12, 111. [Google Scholar] [CrossRef]
- Marchetti, D.; McCutcheon, I.; Ross, M.; Nicolson, G. Inverse expression of neurotrophins and neurotrophin receptors at the invasion front of human-melanoma brain metastases. Int J. Oncol. 1995, 7, 87–94. [Google Scholar] [CrossRef]
- Truzzi, F.; Marconi, A.; Lotti, R.; Dallaglio, K.; French, L.E.; Hempstead, B.L.; Pincelli, C. Neurotrophins and their receptors stimulate melanoma cell proliferation and migration. J. Investig. Dermatol. 2008, 128, 2031–2040. [Google Scholar] [CrossRef]
- Liu, X.; Gao, Y.; Lu, Y.; Zhang, J.; Li, L.; Yin, F. Upregulation of nek2 is associated with drug resistance in ovarian cancer. Oncol Rep. 2014, 31, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zeng, L.; Guan, Y.; Feng, X.; Zhu, Y.; Lu, Y.; Shi, C.; Chen, S.; Xia, J.; Guo, J.; et al. High nek2 confers to poor prognosis and contributes to cisplatin-based chemotherapy resistance in nasopharyngeal carcinoma. J. Cell Biochem. 2019, 120, 3547–3558. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.Y.; Yen, C.J.; Chan, S.H.; Chou, Y.W.; Lee, Y.P.; Bao, C.Y.; Huang, C.J.; Huang, W. Nek2 promotes hepatoma metastasis and serves as biomarker for high recurrence risk after hepatic resection. Ann. Hepatol. 2018, 17, 843–856. [Google Scholar] [CrossRef]
- Huang, J.; Sun, S.G.; Hou, S. Aberrant NEK2 expression might be an independent predictor for poor recurrence-free survival and overall survival of skin cutaneous melanoma. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3694–3702. [Google Scholar] [CrossRef]
- Wen, S.; Liu, Y.; Yang, M.; Yang, K.; Huang, J.; Feng, D. Increased nek2 in hepatocellular carcinoma promotes cancer progression and drug resistance by promoting pp1/akt and wnt activation. Oncol. Rep. 2016, 36, 2193–2199. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W.; Wang, Y.; Huang, X.; Zhang, Z.; Chen, B.; Xie, W.; Li, S.; Shen, S.; Peng, B. Nek2 promotes hepatocellular carcinoma migration and invasion through modulation of the epithelial-mesenchymal transition. Oncol. Rep. 2018, 39, 1023–1033. [Google Scholar] [CrossRef]
- Sankaran, D.; Pakala, S.B.; Nair, V.S.; Sirigiri, D.N.; Cyanam, D.; Ha, N.H.; Li, D.Q.; Santhoshkumar, T.R.; Pillai, M.R.; Kumar, R. Mechanism of mta1 protein overexpression-linked invasion: Mta1 regulation of hyaluronan-mediated motility receptor (hmmr) expression and function. J. Biol. Chem. 2012, 287, 5483–5491. [Google Scholar] [CrossRef] [PubMed]
- Tilghman, J.; Wu, H.; Sang, Y.; Shi, X.; Guerrero-Cazares, H.; Quinones-Hinojosa, A.; Eberhart, C.G.; Laterra, J.; Ying, M. Hmmr maintains the stemness and tumorigenicity of glioblastoma stem-like cells. Cancer Res. 2014, 74, 3168–3179. [Google Scholar] [CrossRef]
- Kauffmann, A.; Rosselli, F.; Lazar, V.; Winnepenninckx, V.; Mansuet-Lupo, A.; Dessen, P.; van den Oord, J.J.; Spatz, A.; Sarasin, A. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene 2008, 27, 565–573. [Google Scholar] [CrossRef]
- Lu, H.; Liu, S.; Zhang, G.; Kwong, L.N.; Zhu, Y.; Miller, J.P.; Hu, Y.; Zhong, W.; Zeng, J.; Wu, L.; et al. Oncogenic braf-mediated melanoma cell invasion. Cell Rep. 2016, 15, 2012–2024. [Google Scholar] [CrossRef]
- Ravindran Menon, D.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Rad, E.B.; et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene 2015, 34, 4448–4459. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.; Pantazopoulou, V.; Johansson, E.; Pietras, A. The p75 neurotrophin receptor enhances hif-dependent signaling in glioma. Exp. Cell Res. 2018, 371, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Anguiano, G.; Lugo-Trampe, J.J.; Camacho, A.; Said-Fernandez, S.; Mercado-Hernandez, R.; Zomosa-Signoret, V.; Rojas-Martinez, A.; Ortiz-Lopez, R. Comparison of specific expression profile in two in vitro hypoxia models. Exp. Ther. Med. 2018, 15, 4777–4784. [Google Scholar] [CrossRef] [PubMed]
- Dusedau, H.P.; Kleveman, J.; Figueiredo, C.A.; Biswas, A.; Steffen, J.; Kliche, S.; Haak, S.; Zagrebelsky, M.; Korte, M.; Dunay, I.R. P75(ntr) regulates brain mononuclear cell function and neuronal structure in toxoplasma infection-induced neuroinflammation. Glia 2019, 67, 193–211. [Google Scholar] [CrossRef]
- Cragnolini, A.B.; Huang, Y.; Gokina, P.; Friedman, W.J. Nerve growth factor attenuates proliferation of astrocytes via the p75 neurotrophin receptor. Glia 2009, 57, 1386–1392. [Google Scholar] [CrossRef]
- Schachtrup, C.; Ryu, J.K.; Mammadzada, K.; Khan, A.S.; Carlton, P.M.; Perez, A.; Christian, F.; le Moan, N.; Vagena, E.; Baeza-Raja, B.; et al. Nuclear pore complex remodeling by p75(ntr) cleavage controls tgf-beta signaling and astrocyte functions. Nat. Neurosci. 2015, 18, 1077–1080. [Google Scholar] [CrossRef]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wolfel, T.; Holzel, M.; et al. Melanomas resist t-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef]
- Guida, M.; Tommasi, S.; Strippoli, S.; Natalicchio, M.I.; de Summa, S.; Pinto, R.; Cramarossa, A.; Albano, A.; Pisconti, S.; Aieta, M.; et al. The search for a melanoma-tailored chemotherapy in the new era of personalized therapy: A phase ii study of chemo-modulating temozolomide followed by fotemustine and a cooperative study of goim (gruppo oncologico italia meridionale). BMC Cancer 2018, 18, 552. [Google Scholar] [CrossRef]
- Ngo, M.; Han, A.; Lakatos, A.; Sahoo, D.; Hachey, S.J.; Weiskopf, K.; Beck, A.H.; Weissman, I.L.; Boiko, A.D. Antibody therapy targeting cd47 and cd271 effectively suppresses melanoma metastasis in patient-derived xenografts. Cell Rep. 2016, 16, 1701–1716. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell rna-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Dobbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Foppen , M.H.; et al. Low mitf/axl ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Chakravarti, N.; Aardalen, K.; Lazar, A.J.; Tetzlaff, M.T.; Wubbenhorst, B.; Kim, S.B.; Kopetz, S.; Ledoux, A.A.; Gopal, Y.N.; et al. Molecular profiling of patient-matched brain and extracranial melanoma metastases implicates the pi3k pathway as a therapeutic target. Clin. Cancer Res. 2014, 20, 5537–5546. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Widmer, D.S.; Hoek, K.S.; Cheng, P.F.; Eichhoff, O.M.; Biedermann, T.; Raaijmakers, M.I.G.; Hemmi, S.; Dummer, R.; Levesque, M.P. Hypoxia contributes to melanoma heterogeneity by triggering hif1alpha-dependent phenotype switching. J. Investig. Dermatol. 2013, 133, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Rappa, G.; Fodstad, O.; Lorico, A. The stem cell-associated antigen cd133 (prominin-1) is a molecular therapeutic target for metastatic melanoma. Stem Cells 2008, 26, 3008–3017. [Google Scholar] [CrossRef]
- Guo, R.; Fierro-Fine, A.; Goddard, L.; Russell, M.; Chen, J.; Liu, C.Z.; Fung, K.M.; Hassell, L.A. Increased expression of melanoma stem cell marker cd271 in metastatic melanoma to the brain. Int. J. Clin. Exp. Pathol. 2014, 7, 8947–8951. [Google Scholar]
- Podlesniy, P.; Kichev, A.; Pedraza, C.; Saurat, J.; Encinas, M.; Perez, B.; Ferrer, I.; Espinet, C. Pro-ngf from alzheimer’s disease and normal human brain displays distinctive abilities to induce processing and nuclear translocation of intracellular domain of p75ntr and apoptosis. Am. J. Pathol. 2006, 169, 119–131. [Google Scholar] [CrossRef]
- Bilderback, T.R.; Grigsby, R.J.; Dobrowsky, R.T. Association of p75(ntr) with caveolin and localization of neurotrophin-induced sphingomyelin hydrolysis to caveolae. J. Biol. Chem. 1997, 272, 10922–10927. [Google Scholar] [CrossRef]
- Ceni, C.; Kommaddi, R.P.; Thomas, R.; Vereker, E.; Liu, X.; McPherson, P.S.; Ritter, B.; Barker, P.A. The p75ntr intracellular domain generated by neurotrophin-induced receptor cleavage potentiates trk signaling. J. Cell Sci. 2010, 123, 2299–2307. [Google Scholar] [CrossRef]
- van Strien, M.E.; Sluijs, J.A.; Reynolds, B.A.; Steindler, D.A.; Aronica, E.; Hol, E.M. Isolation of neural progenitor cells from the human adult subventricular zone based on expression of the cell surface marker cd271. Stem Cells Transl. Med. 2014, 3, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Young, K.M.; Merson, T.D.; Sotthibundhu, A.; Coulson, E.J.; Bartlett, P.F. P75 neurotrophin receptor expression defines a population of bdnf-responsive neurogenic precursor cells. J. Neurosci. 2007, 27, 5146–5155. [Google Scholar] [CrossRef] [PubMed]
- Meeker, R.B.; Williams, K.S. The p75 neurotrophin receptor: At the crossroad of neural repair and death. Neural. Regen. Res. 2015, 10, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a020420. [Google Scholar] [CrossRef]
- Kisiswa, L.; Fernandez-Suarez, D.; Sergaki, M.C.; Ibanez, C.F. Rip2 gates traf6 interaction with death receptor p75(ntr) to regulate cerebellar granule neuron survival. Cell Rep. 2018, 24, 1013–1024. [Google Scholar] [CrossRef]
- Meeker, R.; Williams, K. Dynamic nature of the p75 neurotrophin receptor in response to injury and disease. J. Neuroimmune Pharmacol. 2014, 9, 615–628. [Google Scholar] [CrossRef]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of cell survival by secreted proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef]
- Nykjaer, A.; Willnow, T.E. Sortilin: A receptor to regulate neuronal viability and function. Trends Neurosci. 2012, 35, 261–270. [Google Scholar] [CrossRef]
- Fujita, Y.; Yamashita, T. Axon growth inhibition by rhoa/rock in the central nervous system. Front. Neurosci. 2014, 8, 338. [Google Scholar] [CrossRef]
- Clucas, J.; Valderrama, F. Erm proteins in cancer progression. J. Cell Sci. 2014, 127, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Barker, P.A. Neurotrophin signaling through the p75 neurotrophin receptor. Prog. Neurobiol. 2002, 67, 203–233. [Google Scholar] [CrossRef]
- Maguire, L.H.; Thomas, A.R.; Goldstein, A.M. Tumors of the neural crest: Common themes in development and cancer. Dev. Dyn. 2015, 244, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Shonukan, O.; Bagayogo, I.; McCrea, P.; Chao, M.; Hempstead, B. Neurotrophin-induced melanoma cell migration is mediated through the actin-bundling protein fascin. Oncogene 2003, 22, 3616–3623. [Google Scholar] [CrossRef]
- Marchetti, D.; McQuillan, D.J.; Spohn, W.C.; Carson, D.D.; Nicolson, G.L. Neurotrophin stimulation of human melanoma cell invasion: Selected enhancement of heparanase activity and heparanase degradation of specific heparan sulfate subpopulations. Cancer Res. 1996, 56, 2856–2863. [Google Scholar] [CrossRef]
- Walch, T.E.; Albino, A.P.; Marchetti, D. Correlation of overexpression of the low-affinity p75 neurotrophin receptor with augmented invasion and heparanase production in human malignant melanoma cells. Int J. Cancer 1999, 82, 112–120. [Google Scholar] [CrossRef]
- Yamada, K.; Nomura, N.; Yamano, A.; Yamada, Y.; Wakamatsu, N. Identification and characterization of splicing variants of plekha5 (plekha5) during brain development. Gene 2012, 492, 270–275. [Google Scholar] [CrossRef]
- Bao, X.; Shi, J.; Xie, F.; Liu, Z.; Yu, J.; Chen, W.; Zhang, Z.; Xu, Q. Proteolytic release of the p75(ntr) intracellular domain by adam10 promotes metastasis and resistance to anoikis. Cancer Res. 2018, 78, 2262–2276. [Google Scholar] [CrossRef]
- Verbeke, S.; Tomellini, E.; Dhamani, F.; Meignan, S.; Adriaenssens, E.; Xuefen le, B. Extracellular cleavage of the p75 neurotrophin receptor is implicated in its pro-survival effect in breast cancer cells. FEBS Lett. 2013, 587, 2591–2596. [Google Scholar] [CrossRef]
- Becker, K.; Cana, A.; Baumgartner, W.; Spitzbarth, I. P75 neurotrophin receptor: A double-edged sword in pathology and regeneration of the central nervous system. Vet. Pathol. 2018, 55, 786–801. [Google Scholar] [CrossRef]
- Johnston, A.L.; Lun, X.; Rahn, J.J.; Liacini, A.; Wang, L.; Hamilton, M.G.; Parney, I.F.; Hempstead, B.L.; Robbins, S.M.; Forsyth, P.A.; et al. The p75 neurotrophin receptor is a central regulator of glioma invasion. PLoS Biol. 2007, 5, e212. [Google Scholar] [CrossRef] [PubMed]
- Wrensch, M.; Minn, Y.; Chew, T.; Bondy, M.; Berger, M.S. Epidemiology of primary brain tumors: Current concepts and review of the literature. Neuro. Oncol. 2002, 4, 278–299. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.Y.; Saldanha-Gama, R.F.; Rahn, J.J.; Hao, X.; Zhang, J.; Dang, N.H.; Alshehri, M.; Robbins, S.M.; Senger, D.L. Glioma invasion mediated by the p75 neurotrophin receptor (p75(ntr)/cd271) requires regulated interaction with pdlim1. Oncogene 2016, 35, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, H.; Yamashita, T.; Yoshikawa, H.; Tohyama, M. Pka phosphorylates the p75 receptor and regulates its localization to lipid rafts. EMBO J. 2003, 22, 1790–1800. [Google Scholar] [CrossRef]
- Chen, C.; Shin, J.H.; Eggold, J.T.; Chung, M.K.; Zhang, L.H.; Lee, J.; Sunwoo, J.B. Esm1 mediates ngfr-induced invasion and metastasis in murine oral squamous cell carcinoma. Oncotarget 2016, 7, 70738–70749. [Google Scholar] [CrossRef]
- Chung, M.K.; Jung, Y.H.; Lee, J.K.; Cho, S.Y.; Murillo-Sauca, O.; Uppaluri, R.; Shin, J.H.; Sunwoo, J.B. Cd271 confers an invasive and metastatic phenotype of head and neck squamous cell carcinoma through the upregulation of slug. Clin. Cancer Res. 2018, 24, 674–683. [Google Scholar] [CrossRef]
- Mochizuki, M.; Tamai, K.; Imai, T.; Sugawara, S.; Ogama, N.; Nakamura, M.; Matsuura, K.; Yamaguchi, K.; Satoh, K.; Sato, I.; et al. Cd271 regulates the proliferation and motility of hypopharyngeal cancer cells. Sci. Rep. 2016, 6, 30707. [Google Scholar] [CrossRef]
- Mochizuki, M.; Nakamura, M.; Sibuya, R.; Okazaki, T.; Abe, J.; Nakagawa, T.; Takahashi, S.; Yamazaki, T.; Imai, T.; Takano, A.; et al. Cd271 is a negative prognostic factor and essential for cell proliferation in lung squamous cell carcinoma. Lab. Investig. 2019, 99, 1349–1362. [Google Scholar] [CrossRef]
- Morita, S.; Mochizuki, M.; Wada, K.; Shibuya, R.; Nakamura, M.; Yamaguchi, K.; Yamazaki, T.; Imai, T.; Asada, Y.; Matsuura, K.; et al. Humanized anti-cd271 monoclonal antibody exerts an anti-tumor effect by depleting cancer stem cells. Cancer Lett. 2019, 461, 144–152. [Google Scholar] [CrossRef]
- Krygier, S.; Djakiew, D. Neurotrophin receptor p75(ntr) suppresses growth and nerve growth factor-mediated metastasis of human prostate cancer cells. Int. J. Cancer 2002, 98, 1–7. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using t-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using t-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]