Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Vivek Singh and Version 2 by Peter Tang.
The process of corneal wound healing is complex and induces scar formation. Corneal scarring is a leading cause of blindness worldwide. The fibrotic healing of a major ocular wound disrupts the highly organized fibrillar collagen arrangement of the corneal stroma, rendering it opaque. The process of regaining this organized extracellular matrix (ECM) arrangement of the stromal layer to restore corneal transparency is complicated. The surface retention capacity of ocular drugs is poor, and there is a large gap between suitable corneal donors and clinical requirements.
- corneal scar
- molecular therapeutics
- exosome
- si/microRNAs
- recombinant viral vectors
- bioactive molecules
- nanotechnology
1. Introduction
Unregulated tissue fibrosis is generally associated with the development of chronic diseases. Ocular insults to the cornea, such as alkali burns, injuries, and inflammation, trigger the complex process of wound healing to maintain its structural integrity, functional integrity, and transparency [1]. This is mainly characterized by the migration and proliferation of corneal epithelial cells, followed by the differentiation of stromal keratocytes into myofibroblasts, the excess deposition of extracellular matrix (ECM) components, an increase in alpha-smooth muscle actin (α-SMA) expression, and ECM remodeling, resulting in corneal opacity [2][3][2,3]. The accumulation of these events leads to a scarred cornea (Figure 1). Corneal scarring is the fourth most common cause of blindness, affecting approximately five million people globally each year [4]. A scarred cornea is classified according to its degree of opacity. Nebular opacity is caused by superficial scars on the ocular surface. As the severity of corneal scarring increases, the degree of opacity progresses from nebular to macular and leucoma [5]. Superficial scars heal over a period of three months, restoring vision; however, deep scars generally worsen, leading to blindness [6].
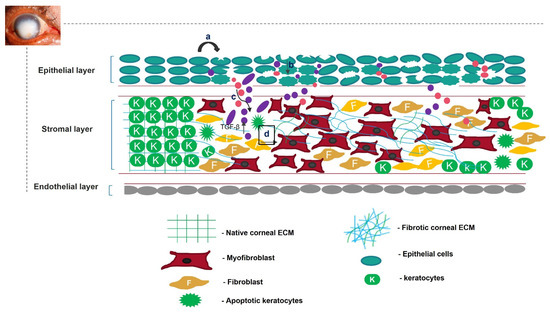
Figure 1. Overview of corneal wound-healing process and scar formation. After an ocular wound, specifically epithelial wounding, (a) the epithelial cells undergo apoptosis, (b) and epithelial cell markers are lost because of corneal epithelial injury, triggering their migration and proliferation. A deep injury disrupts Bowman’s membrane, allowing the invasion of neutrophils (pink) and macrophages (purple) into the stromal layer and releasing TGF-β. This increases the concentration of TGF-β in the stromal layer (c). Therefore, the keratocytes undergo apoptosis, differentiate into myofibroblasts, and disrupt the arrangement of the corneal ECM (d).
Finding an appropriate therapeutic technique to heal a scarred cornea efficiently has proven difficult. Corneal transplantation is an unsuitable therapy for treating scarring because of the lack of appropriate corneal donors [7]. The use of the amniotic membrane as an ocular bandage because of its anti-angiogenic and antifibrotic properties is a popular treatment method. However, its use is limited because of ethical problems related to tissue donation, potential risks of the transmission of diseases, inter-amnion variability, and reproducibility [8]. Topical ocular drops are a simple way of treating a scarred cornea. However, their low bioavailability and rapid drainage from the ocular surface limit their popularity [9].
2. Scarring of Cornea
The cornea is a structurally and functionally integrated avascular organ with three cellular layers interrupted by two acellular layers [10]. It is resistant to minor wounds; however, major ocular trauma can compromise its transparency [11]. A significant abrasion might disrupt the basement and Bowman’s membranes, leading to interactions between the epithelial and stromal layers of the cornea. This interaction initiates the complex wound-healing process [12][13][12,13]. Various growth factors, such as fibroblast growth factor (FGF), epidermal growth factor (EGF), and IGF-1, cross the disrupted Bowman’s membrane from the tear film to reach the stromal layer of the cornea during the initial wound-healing process [14][15][14,15]. In the epithelial layer, wounded epithelial cells undergo apoptosis, migration, and proliferation. The concentration of interleukin-1 (IL-1) increases in the epithelial layer upon injury. Therefore, IL-1 crosses the disrupted Bowman’s membrane into the stromal layer and becomes a major factor in killing keratocytes [16]. In the stromal layer, the corneal injury increases the expression of keratocyte membrane-bound αVβ/αβ integrins [17] and various metalloproteases, such as matrix metalloprotease 2 (MMP2) or MMP9 [18]. This cumulatively activates TGF-β from latency [19][20][19,20]. Neutrophils, monocytes, and macrophages infiltrate the wounded area during the healing process. They either become incorporated into fibrous DNA-like structures known as neutrophil extracellular traps of ECM or secrete TGF-β, increasing the concentration of TGF-β in the stromal layer [21][22][23][24][21,22,23,24]. Therefore, TGF-β takes over the control of the corneal wound-healing process. Quiescent keratocytes transdifferentiate into myofibroblasts, enlarged spindle-shaped cells characterized by the neo-expression of α-SMA in the presence of TGF-β. Incorporating α-SMA into the corneal ECM generates a contractile force that disrupts the orthogonal arrangement of its collagen fibrils, altering its normal curvature [25][26][25,26]. The active form of TGF-β first binds to the keratocyte plasma-membrane-bound TGF-β type I/II receptor (TBRI/II) and switches on the downstream SMAD-dependent signaling cascade [27]. This increases the expression of various fibrotic genes, such as MMPs and connective tissue growth factor (CTGF) [28]. In addition to TGF-β, other neuropeptides, such as calcitonin gene-related peptide, vasoactive intestinal peptide, and substance P (SP), are released from the axonal nerve endings of corneal epithelial layers. These neuropeptides cross the disrupted Bowman’s membrane to reach the stromal layer of the cornea. Substance P binds to the neurokinin 1 receptor (NK1R) of keratocytes and activates three different signaling pathways [29]. The SP/NK1R complex activates the RhoA/ROCK pathway to phosphorylate LIM kinase (LIMK). This inactivates cofilin (an actin-cleaving enzyme) [30], preventing actin degradation and promoting actin polymerization. Actin mostly remains bound to myocardin-related transcription factors A/B (MRTFA/B); however, the increased actin polymerization allows myocardin-related transcription factor A/B (MRTFA/B) to bind to serum response factor, activating profibrotic genes, such as MMPs [31][32][31,32]. The SP/NK-IR complex activates phospholipase C-beta and adenylyl cyclase, increasing the intracellular calcium level and cyclic adenosine monophosphate (cAMP) concentrations. This switches on protein kinase A and activates the extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) pathway, increasing the proliferation of keratocytes and fibroblast cells [33][34][33,34]. The increased Ca2+ ions bind to calmodulin and cohere with the Kca3.1 ion channel of keratocytes to hyperpolarize the cells. This causes the cells to skip the G1 phase of the cell cycle, promoting fibrotic cell proliferation [35]. The increase in cellular stress increases the intracellular reactive oxygen species (ROS) levels, which share a positive feedback loop with the secretion of various growth factors, such as TGF-β, proinflammatory cytokines, etc. [36] Interleukin 10 (IL-10), a proinflammatory cytokine, binds to its cognate receptor and activates the nuclear factor kappa B (NF-κβ) pathway, increasing stress and other inflammatory genes’ expression [37]. TGF-β also increases the expression of bromodomain-containing protein 4 (BRD4), increasing the expression of kelch-like ECH-associated protein 1 (keap1). Keap1 might bind to the antioxidant gene nuclear factor erythroid 2–related factor 2 (Nrf-2) and undergo proteasomal degradation [38][39][38,39]. Cellular stress also increases the expression of the ubiquitin-specific peptidase-10 (USP10) gene. The USP10 gene stabilizes p53, increasing apoptosis of wounded keratocytes. However, it also deubiquitinates integrins, contributing to the activation of TGF-β and fibrosis [40]. The apoptotic cells also release TGF-β and other inflammatory cytokines, completing the loop of fibrosis [41][42][41,42] (Figure 2).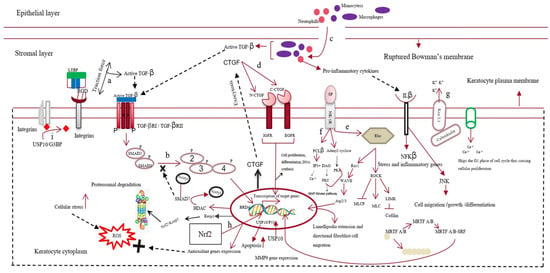
Figure 2. Mechanism of corneal scarring. This figure represents the keratocytes of the stromal layer, the epithelial layer, the disrupted Bowman’s membrane, and the stromal layer of the human cornea. The mechanism of corneal scarring is depicted in a single keratocyte cell for simplicity; (a) activation of transforming growth factor-β (TGF-β) from its latent state; (b) SMAD-dependent TGF-β signaling pathway; (c) infiltration of neutrophils and macrophages into the stromal layer and their secretion of active TGF-β; (d) CTGF (connective tissue growth factor) signaling pathway; (e) substance-P-mediated activation of the Rho/ROCK signaling cascade, leading to actin polymerization and MMP gene expression; (f) increase in intracellular Ca level by Substance-P-mediated PCL-β activation; (g) Kca3.1 ion-channel-mediated hyperpolarization of the keratocyte plasma membrane and subsequent influx of Ca ions; (h) Nrf2 (nuclear factor erythroid 2–related factor 2)-mediated antioxidant gene production; however, BRD4 (bromodomain-containing protein 4) increases keap1 expression, leading to the formation of the kelch-like ECH-associated protein 1 (keap1)/Nrf2 complex and their subsequent degradation; (i) deubiquitylation of integrins by the USP10/G3BP complex.
3. Regeneration of Scarless Cornea
Restoring useful vision is the primary concern of patients with corneal scarring. This can be achieved by preventing the excessive migration of corneal ECM components during wound closure. TGF-β, the growth factor responsible for disrupting the corneal ECM, must remain inactive during the wound-healing process.3.1. Exosomes as A Therapeutic Tool
Exosomes are membrane-bound vesicles released by cells into their extracellular space. They have emerged as a novel therapeutic approach over cell-based therapy for treating corneal scarring [47]. Most stem-cell-based therapies face several challenges, such as immunogenicity, the high cost of production, ethical concerns, stability, and survival. However, exosomes have less stringent safety and regulatory requirements than other stem-cell-based therapies. These microvesicles act as biological vehicles that deliver parent cell cargo to the target cells. The exosomes are internalized by the target cells by either receptor-mediated endocytosis or simple membrane fusion. Upon reaching the cytoplasm of the target cell, the exosomes release their contents, activating various signaling cascades [48]. Exosomes can be isolated from stem cells using simple techniques, such as ultracentrifugation, ultrafiltration, size exclusion chromatography, and immunity capture. These vesicles can deliver antifibrotic proteins and miRNAs from stem cells to the ocular surface to modulate the therapeutic signaling cascade of the scarred cornea. An increase in the concentration of chemokines and cytokines at the injury site is the hallmark of wound healing. Interleukin 8 (IL-8), a proinflammatory cytokine, binds to glycosaminoglycans (GAGs). In this GAG-bound form, IL-8 interacts with the C-X-C motif chemokine receptor 1 (CXCR1) and CXCR2 of neutrophils that infiltrate at the site of injury, leading to oligomerization and a haptotactic gradient. This haptotactic gradient directs the migration of neutrophils to the wounded area and seeds the process of corneal fibrosis [49]. Corneal stromal stem cells (CSSCs) express tumor necrosis factor (TNF)-stimulated gene 6 (TSG-6), a hyaluronan-binding protein, during inflammation [50]. This 35 kDa protein interacts with IL-8 via its link module domain and disrupts the binding of IL-8 and GAGs, preventing neutrophil migration [51][52][51,52]. Shojaati et al. [53] isolated extracellular vesicles (EVs) from CSSCs, mixed them with fibrin gel, and administered the EV–fibrin gel to the surface of an eye with corneal debridement. After 2 weeks of debridement, the EV-treated cornea showed minor corneal scarring compared to CSSC-treated and untreated controls. EVs were more effective in preventing neutrophil infiltration, reducing fibrotic marker expression, and restoring corneal morphology. Cultured CSSCs treated with exosomes isolated from adipose-derived stem cells (ADSCs) showed optimal proliferation, reduced apoptosis, increased aldehyde dehydrogenase 1 (ALDH1), decreased MMP1, MMP3, and MMP9 expression, and increased collagen I, II, III, IV, and V expression compared to untreated CSC [54]. Therefore, ADSC-derived exosomes might revive the plasticity of transformed corneal keratocytes or stromal cells by increasing keratocyte marker expression and ECM production. The therapeutic effects of ADSCs and ADSC-derived exosomes could be attributed to microRNA-19a, which binds the 3′UTR of homeodomain-containing serine/threonine kinase 2 (HIPK2). MicroRNA-19a post-transcriptionally suppresses homeodomain-interacting protein kinase 2 (HIPK2), preventing the activation of the Jun N-terminal kinase (JNK) pathway and fibrosis [55]. Corneal keratocytes, when co-cultured with ADSC-derived exosomes, reduce HIPK2, phosphorylated SMAD3, and p53 expression, preventing the transformation of keratocytes into myofibroblasts and apoptosis of the nearby injured cells. This condition was reversed by the lentivirus-mediated overexpression of HIPK2 in keratocytes, confirming the role of miRNA-19a in corneal scar healing [56]. Exosome-based therapy is still in its nascency and requires preclinical research to be used for treating scars. Exosomes isolated from corneal stromal stem cells effectively prevent corneal scarring and require more preclinical safety and toxicity studies. Similarly, ADSC-derived exosomes reduce apoptosis and restore keratocyte marker expression in corneal stromal cells. Further characterization of these experiments is required to check their preclinical efficacy and gain better molecular insight to understand their therapeutic benefits. Exosomes from HCEs can hasten the corneal wound-healing process and prevent corneal scarring (Figure 3).The parent cells could be engineered to overexpress specific antifibrotic proteins. Exosomes consisting of this protein can be used in combination with tissue-derived microparticles to increase the effectiveness of exosome-based therapy for a scarred cornea.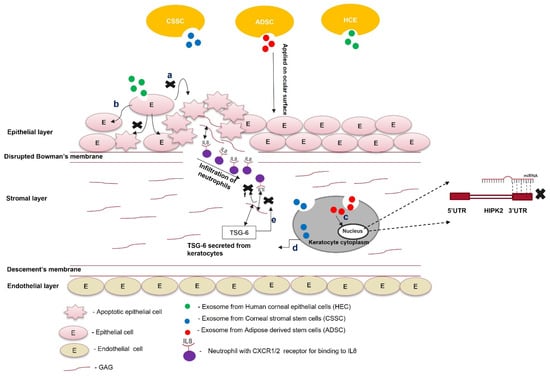
Figure 3. Role of exosomes in the treatment of scars. Exosomes from different cell types (ADSCs, CSSCs, and HECs) are loaded within the fibrin gel or encapsulated in the hydrogel and applied on the ocular surface; (a) exosomes from HECs taken up by the epithelial cells prevent paraptosis (apoptosis caused by hypoxic conditions); (b) epithelial cells treated with exosomes do not undergo apoptosis; (c) exosomes from ADSCs contain miR-19a, which post-transcriptionally silences HIPK2 and attenuates the JNK pathway; (d) exosomes from CSSCs contain TSG-6 protein, and once they are taken up by keratocytes, the TSG-6 proteins are secreted within them; (e) TSG-6 protein binds to GAG via its LINK domain and prevents the binding of IL8 to the GAG of corneal stroma; (f) the unbound form of IL-8 cannot be presented to the CXC1/2 receptor of neutrophils. It attenuates the infiltration of neutrophils to the wounded area of the cornea.
3.2. Targeted Gene Silencing for the Generation of A Scarless Cornea
The idea of directly targeting the upregulated genes of the corneal scarring signaling cascade to heal a scarred cornea has attracted researchers over the past few years. Semaphorin 3A (SEMA-3A), ubiquitin-specific protease-10 (USP 10), and calmodulin/calcium-activated K+ channel 3.1 (Kca3.1) are upregulated during the corneal wound-healing process. The increased expression of these genes contributes to corneal fibrosis. Therefore, targeting these genes using siRNA can mitigate the scarred corneal environment. The complete knockout of a targeted gene is undesirable because genetically modifying a mammalian cell is difficult, time-consuming, and expensive; moreover, knocking out a gene could have lethal consequences. Therefore, the in vivo delivery of small oligonucleotide sequences, designed to target certain mRNAs, using a suitable delivery vehicle to silence a target gene might be an effective way to prevent corneal fibrosis.3.3. Protein Overexpression in Preventing Corneal Scarring
The cornea is an ideal tissue for gene therapy, as the ocular surface can be an excellent platform for the topical application of various viral/non-viral gene delivery vectors. The overexpression of specific genes in corneal epithelial or stromal cells that block the TGF-β signaling cascade can prevent scar formation.3.4. MicroRNA Therapies in Regenerating Cornea without Scars
MicroRNAs are 21-nucleotide-long, non-coding RNAs that can post-transcriptionally silence the expression of their target genes. These microRNAs form an RNA-induced silencing complex (RISC) with Argonaute proteins. These proteins guide them towards the target mRNA, where they pair with the target mRNA and silence its expression [57][87]. MicroRNAs in the cornea affect various cellular processes, such as cell migration, differentiation, proliferation, and metabolism. Any dysregulation in miRNA levels during corneal injury can lead to corneal neovascularization and scar formation. An J et al. [58][88] observed a sharp decrease in the expression of miR-204 in a murine corneal wound model. MicroRNA-204 is abundant in the cornea. It inhibits corneal cell proliferation by G1 phase arrest. Therefore, low miR-204 levels during corneal wounds account for the excessive proliferation of transformed epithelial cells, myofibroblasts, ECM remodeling, and fibrosis during quick wound healing. Wang Y et al. [59][89] compared lens epithelial cells from healthy donors and patients with posterior capsular opacification (PCO). They showed increased α-SMA and vimentin and decreased E-cadherin in the diseased cells, indicating opacification. An in vitro PCO model was developed and transfected with miR-204. The lens epithelium cells showed increased E-cadherin and decreased α-SMA compared to non-transfected controls and miR-204-5p-inhibitor-transfected cells. MicroRNA-204 prevents TGF-β-mediated EMT and fibrosis by targeting SMAD4, disrupting the SMAD2/3-SMAD4 complex. It also decreases the expression of the Hey/HMGA transcription factors, which induce EMT through CDH1 repression and SNAIL activation [60][61][90,91]. A subconjunctival injection of a recombinant adeno-associated vector (rAAV-miR-204) prevented neovascularization in murine alkali-burned corneas, indicating the anti-angiogenic role of miR-204 [62][92]. The downregulation of miR-145 and the upregulation of miR-204 and miR-133b can prevent TGF-β-mediated corneal fibrosis. MicroRNA-based gene targeting is a promising therapeutic approach to prevent and heal corneal scarring (Figure 4).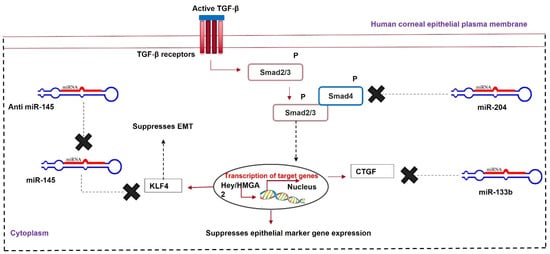
Figure 4. MicroRNA therapy for corneal scarring. Upregulation of miR-204 and miR-133b and downregulation of miR-145 help in healing a scarred cornea via modulating Krüppel-like factor 4 (KLF4), Smad2/3, and CTGF.
3.5. Bioactive Molecules as HDAC Inhibitors in the Regeneration of Scarless Cornea
The wound-healing process of an injured cornea involves three overlapping phases: the inflammatory phase, the proliferative or fibrotic phase, and the final remodeling phase [63][100]. These events change the native morphology of the corneal stromal ECM, rendering it opaque. During the initial inflammatory phase of the healing process, corneal cells exhibit high levels of proinflammatory cytokines. During the initial phase of wound healing, the infiltrated neutrophils, monocytes, or activated macrophages secrete TGF-β, increasing its local concentration around the wounded area. TGF-β guides the transition of the wound from the inflammatory phase to the fibrotic phase by decreasing the local proinflammatory cytokine level. The timely termination of the initial inflammatory phase is required for the cells to enter the cell proliferation and ECM synthesis phase, healing the wound [64][101]. A lesser-known pathway that TGF-β uses for this transition is by modulating histone acetylation. TGF-β recruits histone deacetylases (HDACs), removing the acetyl group from the lysine residues of histones H3 and H4 of anti-inflammatory genes. Histone acetylation is associated with anti-inflammatory gene activation; therefore, HDAC is recruited in the presence of TGF-β, deactivating various anti-inflammatory genes and enabling the transition to the fibrotic phase [65][66][67][68][69][102,103,104,105,106]. Trichostatin A (TSA), a deacetylase inhibitor, prevents the deacetylation of histone H3 or H4 of anti-inflammatory genes. TSA prevented the TGF-β-mediated transformation of corneal fibroblasts to myofibroblasts in vitro and also reduced the corneal opacity in a rabbit model of corneal haze [70][71][107,108]. A possible mechanism of action for HDAC inhibitors is increasing the local concentration of TGF-β during wounding, leading to HDAC recruitment, SMAD7 deacetylation, SMAD7 ubiquitinylation, and proteasomal degradation. This paves the way for TGF-β-mediated corneal scarring. Similarly, suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor, reduced α-SMA and MMP9 expression in equine corneal fibroblasts treated with TGF-β. This shows that SAHA can prevent TGF-β-mediated corneal fibrosis [72][109]. Epigenetic modulators, such as TSA and SAHA, might be useful bioactive molecules for treating corneal scars.3.6. Guided Wound Healing to Prevent Scarring
Corneal wound healing is accompanied by the secretion of various growth factors. During wound healing, these growth factors interact with their cognate receptors present on the surface of the cell to regulate the synthesis of collagen, proteoglycan, and other ECM components’ deposition. Various growth factors, such as IGF-1, platelet-derived growth factor (PDGF), TGF-β, and fibroblast growth factor 1 (FGF-1), either reach the wound site from the tear film or are secreted by the surrounding wounded or apoptotic epithelial cells. Etheredge et al. [73][110] reported that FGF-1 stimulates keratocyte proliferation; however, it inhibits the synthesis of type 1 procollagen and keratan sulfate. Type 1 procollagen is an essential component of the stromal ECM, and keratan sulfate directs collagen assembly to maintain corneal transparency. FGF-1 induces the formation of hypercellular keratocytes with densely packed cells in the sparse matrix. IGF-1 turns these hypercellular keratocytes into collagenous keratocytes that secrete ECM components, similar to native keratocytes. Therefore, the IGF-1/IGF-receptor signaling pathway prevents the transformation of keratocytes into myofibroblasts and ECM remodeling (Figure 5). However, this is not a predominant signaling pathway during wound healing because the local concentration of TGF-β in the wound microenvironment is high. Therefore, the TGF-β/TGF-βRI pathway takes charge, and TGF-β binds to its receptor on keratocytes to synthesize biglycan, fibronectin containing extra domain A, and hyaluronan. This proteoglycan disrupts the spacing between collagen fibrils, reducing the corneal transparency. It also guides the differentiation of keratocytes into myofibroblasts, characterized by the neo-expression of α-SMA. α-SMA incorporates actin into collagen fibrils and exerts a contractile force on the ECM, disrupting the normal corneal architecture.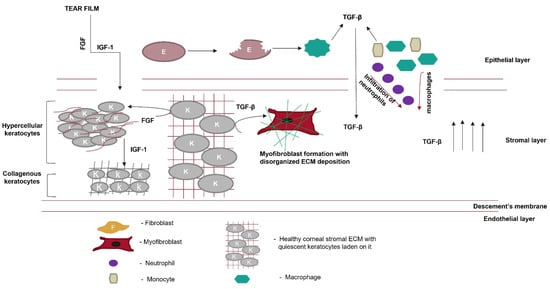
Figure 5. Role of growth factors in scar prevention. Various growth factors, such as fibroblast growth factor (FGF) or insulin-like growth factor (IGF-1), are secreted from the tear film. These growth factors cross the disrupted Bowman’s membrane to reach the corneal stromal layer. FGF converts keratocytes to hypercellular keratocytes. IGF-1 converts hypercellular keratocytes to collagenous keratocytes, which secrete ECM components similarly to the native cornea. Meanwhile, in the epithelial layer, after wounding, epithelial cells undergo apoptosis, and the apoptotic epithelial cells release TGF-β. Neutrophils, macrophages, and monocytes, which migrate from the epithelial layer to the stromal layer, secrete TGF-β, thereby increasing its local concentration. Keratocytes in the presence of TGF-β are converted to myofibroblasts, which disrupt the highly organized fibrillar arrangement of the corneal ECM.
3.7. Clinical Therapy for Scar Prevention
A gold-standard technique to prevent corneal scarring is still under research. Various biomolecules, such as decorin, TPCA-1, glucosamine, acetylcholine, and chitosan, have the potential to heal a scarred cornea via their respective biological pathways. Chen et al. [74][113] reported the significance of glucosamine in corneal scar treatment. This amino sugar finds clinical application in osteoarthritis management [75][114]. However, it significantly enhances intraocular pressure [76][115]. Park et al. [77][116] noted that glucosamine attenuated renal fibrosis by downregulating SMAD2 phosphorylation; therefore, a study of glucosamine’s effects on corneal fibrosis can provide new insights into its clinical applications. Chen’s group observed an increase in the expression, stability, and nuclear localization of KLF4 and a decrease in fibrotic gene expression in HCFs treated with glucosamine and TGF-β. The mechanism underlying the glucosamine-mediated increase in KLF4 expression is still under research. However, a study by Wang DF et al. [78][117] reported that glucosamine increases the expression of the phosphorylated form of PTEN (a phosphatase), leading to an increase in the phosphorylated form of KLF4. This increases the P300 histone acetylase activity, mediating H3 histone acetylation [79][80][118,119]. Glucosamine can form O-linked N-acetylglucosamine via glucosamine transferase activity and the post-translational modification of the proteasome complex to prevent KLF4 degradation [81][120]. Moreover, KLF4 interacts with TGF-β control elements and prevents a proinflammatory environment [82][121]. Therefore, glucosamine might find clinical applications in healing corneal scarring (Figure 6).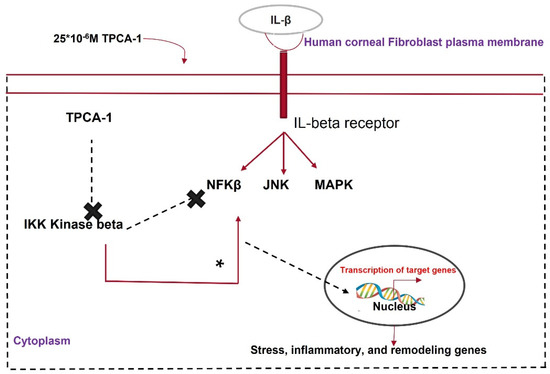
Figure 6.
Mechanism of action of TPCA1 in attenuating corneal fibrosis.
3.8. Nanomedicine in Corneal Scarring Treatment
Nanomedicine is an emerging field in medical science with the potential to revolutionize corneal fibrosis therapy (Figure 7). Nanomedicine utilizes materials with nanometer dimensions to target living cells at the genetic and molecular levels. It ensures efficient and sustained drug release, increasing the bioavailability of drugs. This is especially good for topical eye drops, which have a bioavailability of only 5% [112][152].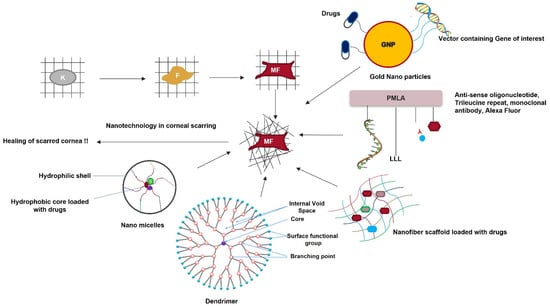
Figure 7. Nanotechnology as a therapeutic tool for reviving scarred corneas. Keratocytes transform into fibroblasts and then into myofibroblasts in the presence of TGF-β. This disrupts the highly organized fibrillar arrangement of the corneal stromal ECM. Nanoparticles loaded with the gene of interest or specific drugs, nanofiber scaffolds loaded with drugs, dendrimers, nanomicelles, and nanopolymers are a few nanomedical approaches for treating a scarred cornea.