1. Indirect Detection
Indirect detection is based on virus-induced morphological changes to host cells or membranes.
1.1. Monkeypox Diagnosis Based on Virus Culture
Some viruses can induce macroscopic lesions (called pocks) on the chick chorioallantoic membrane (CAM). The pattern of pock formation, the time required for pock formation, and the size of the pock have been explored to differentiate different poxvirus infections, including
monkeypox virus (MPXV
) [1][2][27,28]. The morphological changes can be observed with a microscope or the naked eye. For instance, when CAM was inoculated with MPXV, the pocks were visible and could be reckoned with the naked eye
[2][28]. However, detection solely based on the above-mentioned characteristics may not be sufficient for an accurate diagnosis due to overlapping signs and symptoms with other diseases.
Monkeypox isolates are grown in RK13 cells
[3][29], where cytopathic effects are observed within 24–48 h of infection. The major drawback of culture-based virus diagnosis is the prolonged assay time
[4][30], which is not suitable for mass testing scenarios. Further, virus culture methods need biosafety level 3 (BSL3) labs and pose a risk of laboratory-acquired infections
[5][31]. Shell vial culture (SVC) has been developed as an alternative culture method for the rapid in vitro detection of MPXV and other viruses
[4][30]. In this method, a cell monolayer is grown on a cover slip in a shell vial culture tube, and the specimen is inoculated on the monolayer, followed by low-speed centrifugation and immunofluorescence-based detection. The low-speed centrifugation step is introduced to enhance the virus’s infectivity. The mechanical force resulting from low-speed centrifugation is thought to cause cell trauma, which subsequently enhances viral entry into cells, resulting in a reduced cell infection time
[6][32].
1.2. Diagnosis Based on Image Analysis
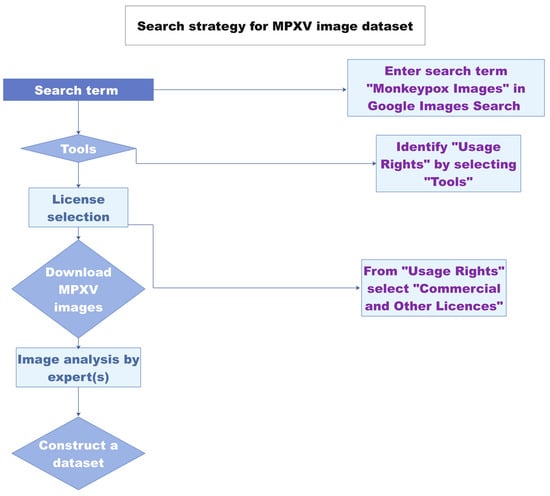
Image digitalization has already gained momentum for infectious disease diagnosis and monitoring. Chatbots have been developed for disease diagnostic evaluation and the recommendation of immediate measures in case a patient contracts SARS-CoV-2
[7][33]. A monkeypox image dataset was constructed comprising 43 original images and 587 images obtained after data augmentation
[8][34] (
Figure 1). Using the newly developed “Monkeypox 2022” dataset, an image classification model was proposed
[9][35]. The study paves the way towards the development of image-analysis-based tools for monkeypox virus detection. The images used in the dataset are from previous outbreaks. The classic MPXV cases were characterized by a generalized rash. In contrast, most of the cases in the current outbreak have localized lesions in anogenital and genitourinary areas
[10][11][36,37]. Since many recent MPXV-infected cutaneous images have been reported, the updated dataset may have added value to the above MPXV 2022 image dataset.
Figure 1. Schematic of the image-based MPXV detection workflow. Redrawn from Ref.
[9][35] with permission from the author. The source content is licensed under a Creative Commons Attributions 4.0 international license.
2. Direct Detection
In the case of direct detection, nucleic acid and protein components of the virus are detected without the need for a pathogen culture. Molecular detection, immunodiagnostics, and sequencing are widely explored direct detection approaches.
2.1. Monkeypox Immunodiagnostics
The hemagglutination test is a simple and cost-effective approach for virus detection. The test is based on the agglutination of erythrocytes in the presence of a virus
[4][30]. The hemagglutination mechanism led to the development of another assay called the hemagglutination inhibition (HI) assay
[12][38]. The HI approach relies on virus-specific antibodies to detect viral antigens. The MPXV strains are tested using hemagglutination and HI tests
[2][28]. The test cannot differentiate MPXV from the variola and vaccinia viruses but can differentiate cowpox from MPXV and can be used to estimate the evolutionary relationships of viral strains or species.
The enzyme-linked immunosorbent assay is a widely used protein detection method
[13][39]. A commercially available Orthopox BioThreat
® Alert Assay for orthopox virus (OPV) detection is a reliable OPV detection method
[14][40]. This antibody-based lateral flow assay captures virus antigens and detects the viral load at 10
4 PFU/mL. The surface protein A27 was found to be the most immunogenic protein for virus particle capture and detection
[15][41]. After a comprehensive screening of A27-binding antibodies, an ELISA approach was developed for orthopoxviruses, including MPXV. The method’s detection limit is 1× 10
3 PFU/mL. Recently, Ulaeto et al. described the characteristics of an LFA for the detection of orthopoxviruses
[16][44]. The assay detects vaccinia virus samples spiked in human saliva and clinical sample buffer with a detection limit of between 10
4 and 10
5 PFU/mL within 20 min. Since this assay detects orthopoxviruses, the test can be further explored for MPXV detection in real samples. Combining the clinical presentation of MPXV with the LFA test could provide a rapid MPXV detection tool. All of the above-mentioned immunodetection modalities are suitable for generic orthopox virus detection applications, but none of them are specific for MPXV.
2.2. Whole-Particle Detection
Finding biomarkers for a newly emerged virus is challenging and may hamper the direct implementation of routine diagnostic methods. In this regard, whole-particle detection using electron microscopy (EM) is a powerful alternative
[17][45]. Transmission electron microscopy is a good first step for the detection of viruses, as it provides information about the shape and amount of viral load with a small sample volume
[18][46]. The use of virus-specific antibodies in immunoelectron microscopy (IEM) further improves the detection accuracy of EM
[19][47]. EM has been used to detect monkeypox and other orthopoxviruses
[20][48]. Although EM is suitable for the laboratory validation of the virus detection results, the approach has certain limitations, such as the high cost of the instrument, the requirement of highly trained staff, and low sample throughput
[20][48].
2.3. Detection by Genome Sequencing
Genome sequencing is the gold standard to identify novel or mutated viruses. Genome sequencing not only identifies the target virus but may pinpoint the presence of other viruses in the sample that can help to create a treatment plan for a particular disease. MPXV detection based on qPCR coupled with genome sequencing has been reported
[21][49]. To date, 200 genome sequences of MPXV isolates from recent outbreaks in non-endemic countries have been reported
[22][50]. Whole-genome sequencing is a time-consuming process and requires expensive instruments, trained staff, and skilled bioinformaticians for computational analyses. These limitations need to be overcome to harness the potential of genome sequencing approaches.
2.4. Monkeypox Virus Detection Based on PCR
The polymerase chain reaction (PCR) is widely regarded as the gold standard for nucleic acid detection. According to WHO recommendations, PCR (conventional or real-time) is a standard method for MPXV laboratory validation
[23][51]. The detection can be combined with sequencing or other orthopox detection assays
[24][19]. Conventional PCR-based MPXV detection involves PCR amplification and restriction digestion of the PCR-amplified fragments to identify MPXV based on restriction fragment length polymorphisms.
A hemagglutinin PCR (HA-PCR) assay was developed based on MPXV-specific primers coupled with
TaqI restriction digestion
[25][52]. The method could not distinguish different MPXV isolates. To improve the detection accuracy of the PCR assay, an A-type inclusion body protein (ATI) gene has been used to detect MPXV and other orthopoxviruses based on PCR-based gene amplification and
XbaI digestion
[26][27][53,54]. The method can differentiate MPXV strains based on restriction digestion. In another development, the open reading frame (ORF) of the ATI gene was identified, sequenced, and compared with other related poxviruses
[28][55]. Unique deletions were found in the OFR of MPXV and were harnessed for the specific detection of the MPXV ATI gene. This PCR method differentiates 19 MPXV strains. The specificity was confirmed by
BglII restriction digestion.
Compared to traditional PCR, real-time PCR is rapid and sensitive. Due to the low GC content and almost 90% genome identity with other Eurasian
orthopoxviruses, designing an MPXV-specific TaqMan assay is challenging. Li et al. developed a real-time PCR assay where minor-groove-binding protein-based (MGB) probes were developed
[29][56]. The use of MGB stabilizes probe–template interactions, enables the use of small probe sequences for single-nucleotide polymorphism (SNP) detection, and enhances assay sensitivity and specificity
[30][57]. The method could detect 15 MPXV isolates at a 10 ng concentration. The assay efficiency with freshly diluted DNA is 97%, while it is reduced to 67% after multiple freeze–thaw cycles. These observations indicate that a fresh sample should be used in order to achieve maximum assay efficiency. The detection of MPXV and other orthopoxviruses based on melting-curve analysis (MCA) has also been reported
[31][32][33][58,59,60]. Both clades (West African and Congo Basin) of MPXV have 99% sequence identity but are significantly different in terms of virulence
[34][61]. It is a big challenge to develop a clade-specific real-time PCR detection approach due to the limited availability of unique sequences.
Multiplex detection can significantly reduce the misidentification of coexisting pathogens
[35][36][63,64]. A multicolor, multiplex approach for MPXV detection was reported where MPXV was specifically detected in the presence of the variola virus (VARV) and the varicella-zoster virus (VZV)
[37][65]. The target genes harboring unique sequences for MPXV, VARV, and VZV are F3L, B12R, and ORF38, respectively. The specificity of the developed approach is 100%, and LODs of 20 copies per reaction for MPXV and VARV and 50 copies per reaction for VZV were reported. The robustness of the approach was demonstrated by successfully detecting the different combinations of MPXV, VARV, and VZV samples.
The standard poxvirus detection approach combines the disease’s clinical symptoms with a generic poxvirus PCR assay, followed by a poxvirus-specific PCR assay
[33][60]. These pan-pox real-time PCR methods are instrumental in the accurate diagnosis of poxvirus infection. Based on the GC content, the chordopoxviruses (poxviruses that infect vertebrates) of the subfamily
Chordopoxvirinae have two distinct genome types: one genome type contains high GC content (>60%), while the other genome type is comprised of low GC content (30–40%)
[38][66]. GC-content-based pan-pox PCR assays have been developed
[38][66]. The assays are termed high-GC PCR and low-GC PCR assays. The developed PCR assays detected DNA samples from more than 150 isolates and strains of chordopoxviruses. The detection approach is based on conventional PCR, and PCR amplicons are evaluated by
TaqI RFLP patterns. In a similar line of work, a real-time PCR assay for the universal detection of orthopoxviruses was reported
[36][64]. The system was reported to be able to detect poxviruses excluded in a previous study
[38][66] as well as those from the subfamily
Entomopoxvirinae. This assay targets a 100 bp highly conserved sequence in the D6R gene of poxviruses. The specificity of the assay for vertebrate samples is 99.8%, while it is 99.7% for arthropod samples. The system is 100% sensitive for vertebrate samples and 86.6% sensitive for arthropod samples. The detection limits are reported to be 100 or 1000 copies per reaction, depending on the poxvirus species.
2.5. Detection Based on Isothermal Amplification
More than ten types of different isothermal amplification methods have been reported and demonstrated for nucleic detection
[39][13]. Loop-mediated isothermal amplification (LAMP) and recombinase polymerase amplification (RPA) are well-explored isothermal nucleic acid amplification based virus detection methods
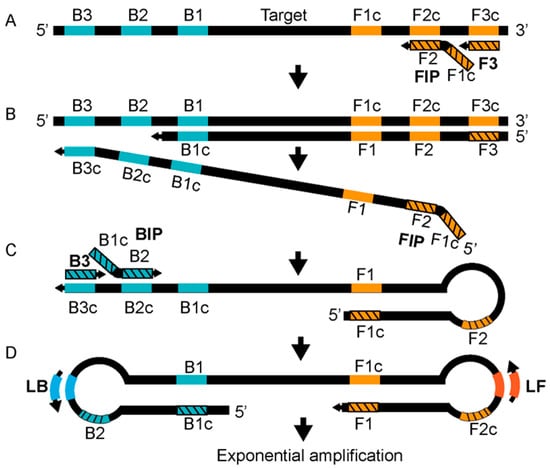
[40][67]. The LAMP technology relies on two internal primers called the forward internal primer (FIP) and the backward internal primer (BIP), two outer primers known as the forward outer primer (F3) and the backward outer primer (B3), and a DNA polymerase with strand displacement activity
[40][67]. The reaction is carried out at 60–65 °C. The amplification reaction is accelerated by using two loop primers, the forward loop (LF) and the backward loop primer (LB)
[41][68]. The annealing of the FIP, which has two target sequences (separated by a spacer) complementary to the two different regions of the template, initiates strand synthesis and elongation (
Figure 2A). Subsequently, the F3 primer displaces the FIP strand, producing a single-stranded DNA (ssDNA) strand that is used as a template by the BIP (
Figure 2B). The BIP, which also has two target sequences complementary to the template DNA at two different regions, starts the strand elongation of the ssDNA template, which is later displaced by the B3 (
Figure 2C). The 5′ and 3′ ends of the template DNA have inward complementary sequences, forming a stem-looped DNA that is exponentially amplified by loop primers (
Figure 2C,D). LAMP-based MPXV-clade-specific assays have been developed where West African (the assay named W-LAMP) and Congo Basin MPXV (the assay named C-LAMP) clades are selectively detected
[42][69]. A turbidimeter is used to analyze the LAMP reaction, and restriction digestion is used to confirm the LAMP products. A LAMP-based method for rapid MPXV detection was recently posted on a preprint server
[43][70]. The assay was developed to detect MPXV clades. The method shows satisfactory sensitivity and response times.
Figure 2. Reaction mechanism of LAMP. See text for details. Redrawn from Becherer et al., 2020, Ref.
[40][67] © The Royal Society of Chemistry 2020, licensed under a Creative Commons Attributions-Noncommercial 3.0 unported license
https://creativecommons.org/licenses/by-nc/3.0/. Accessed on 12 October 2022.
Although promising, the LAMP needs a 60-minute reaction time and six primers. Furthermore, primer design is relatively complex. To overcome these limitations, RPA has been proposed as an attractive alternative
[44][71]. The RPA signal is detected by gel electrophoresis, real-time monitoring
[45][72], or lateral flow assay
[45][72]. In the case of real-time detection, the fluorogenic probe, along with the primers, is added to the reaction system where cleavage of the probe by exonuclease leads to a fluorescent signal. RPA-based MPXV detection shows satisfactory results with reduced assay times and reagent costs
[46][73].
Table 1.
Summary of MPXV diagnostic methods.