Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jiaoyang Jiang and Version 2 by Peter Tang.
The dynamic O-GlcNAc modification of intracellular proteins is an important nutrient sensor for integrating metabolic signals into vast networks of highly coordinated cellular activities. Dysregulation of the sole enzymes responsible for O-GlcNAc cycling, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), and the associated cellular O-GlcNAc profile is a common feature across nearly every cancer type.
- O-GlcNAcylation
- O-GlcNAc transferase (OGT)
- O-GlcNAcase (OGA)
- cancer
- protein–protein interaction (PPI)
1. Introduction
O-linked N-acetylglucosaminylation (O-GlcNAcylation) is an essential post-translational modification (PTM) that dynamically regulates numerous protein functions in response to nutrients and stress [1]. Interestingly, only a single pair of human enzymes maintains the homeostasis of this modification: O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) [2][3][4][5][6][2,3,4,5,6]. OGT transfers the GlcNAc moiety from the sugar donor UDP-GlcNAc to the serine or threonine residues of protein substrates (Figure 1). On the contrary, OGA removes the sugar moiety from O-GlcNAcylated substrates (Figure 1). This reversible O-GlcNAc cycle dynamically modulates protein stability, enzymatic activity, protein–protein interactions (PPIs), and the crosstalk with other types of PTMs [7][8][7,8]. To date, thousands of O-GlcNAcylated proteins have been identified and they play important roles in remarkably diverse cellular processes, including transcription, translation, apoptosis, cell cycle, protein transportation, mitochondrial function, and signal transduction [7][9][10][7,9,10]. Notably, dysregulation of OGT, OGA, and the associated cellular O-GlcNAc profile is commonly detected in all cancers [11]. For instance, upregulated OGT and O-GlcNAcylation are intimately associated with nearly every cancer-related phenotype, ranging from cell proliferation, epithelial–mesenchymal transformation (EMT), angiogenesis, to metastasis [11][12][13][14][11,12,13,14]. Emerging evidence also shows that OGT is involved in regulating/activating cancer stem cell potential and resistance in anti-cancer treatments [14][15][16][14,15,16]. On the other side, both up- and down-regulation of OGA protein levels have been observed in different types and grades of cancer [17][18][19][20][17,18,19,20]. Elevated activity of OGA was also detected in cancer [21]. Furthermore, anti-cancer drugs combined with OGA inhibition using small molecule or genetic approaches have shown synergic inhibitory effects on tumor progression [22][23][24][22,23,24]. More interestingly, a significant correlation between the expression levels of OGT/OGA and the grade/stage of tumors or prognosis has been discovered, promoting mechanistic investigations of these enzymes in cancer [17][25][26][17,25,26]. In general, the abnormal functions of OGT/OGA can make profound impacts on many biological processes, such as metabolic reprogramming, transcription/epigenetic regulation, inflammation, and stress response [27][28][29][30][31][27,28,29,30,31]. These dysregulations, often amplified through a large repertoire of O-GlcNAcylated proteins, fuel cancer malignancies and accelerate disease deterioration. These findings raised significant interest in targeting O-GlcNAc cycling enzymes (OGT and OGA) as a potential new anti-cancer strategy. In the past decade, genetic perturbation and the active-site inhibitors of these two enzymes have been widely used to gain fundamental understanding of their roles in normal and disease conditions, and to evaluate their potential for therapeutic development. Exciting progress has been made; however, significant challenges have also become apparent. One of the main challenges is that OGT and OGA are essential enzymes; prolonged knockdown or knockout of either of them leads to embryonic lethality or deterioration of organ functions [32][33][32,33]. Inhibition of OGT/OGA’s catalytic site brings similar concerns about unpredictable side effects due to the perturbation of global O-GlcNAcylation [34][35][36][37][34,35,36,37]. In addition, the non-catalytic functions of OGT and OGA have been recently reported to regulate cell proliferation and tumor cell growth, respectively, indicating that their active-site inhibition may not be sufficient to halt cancers derived from the aberrant non-catalytic functions of O-GlcNAc cycling enzymes [18][38][18,38]. Hence, there is a critical need to explore new strategies to target OGT/OGA. To develop such new strategies, a better understanding of how OGT and OGA interact with other proteins (e.g., substrates or non-substrate partners) through regions outside of their immediate catalytic sites would be essential. This knowledge will not only aid in defining the malfunctions of OGT/OGA in complex diseases such as cancer, but also facilitate the development of novel strategies to manipulate the interactions of these enzymes and a subset of proteins without global O-GlcNAc perturbation-induced side effects.
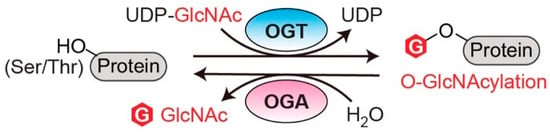
Figure 1. O-GlcNAc cycling enzymes (OGT and OGA) catalyze the reversible protein O-GlcNAcylation. OGT: O-GlcNAc transferase. OGA: O-GlcNAcase. UDP-GlcNAc: uridine diphosphate N-acetylglucosamine.
Perturbed PPIs in cancer (cancer-specific PPIs) is one of the key factors in cancer development [39]. Mapping PPIs has provided invaluable insights into the pathophysiological mechanisms in multiple types of cancer [40]. Moreover, aberrant PPIs are arising as new targets for the development of novel cancer therapy. As many PPI inhibitors have entered clinical trials or applications, this has become an important strategy to impede malignant cancer programming with minimal toxicity [41]. Given the manifold functions of O-GlcNAc cycling enzymes, deciphering their roles from a PPI perspective promises fruitful discoveries and may open new doors for cancer therapeutic interventions. Compared to many other cancer-related proteins (e.g., BCL2, p53, etc.), the protein interactions of OGT/OGA have been significantly less explored, potentially restricted by their transient protein interactions with many O-GlcNAcylated substrates and a lack of a conserved recognition motif [42].
2. Structural Insights of O-GlcNAc Cycling Enzymes as Potential Multi-Interface Hubs for Regulating Complex PPI Networks
Analyses using interdisciplinary approaches, including structure, bioinformatics, and multi-omics, have greatly accelerated our understanding of cancer-specific PPI interface properties and topological features. For example, intrinsically disordered regions (IDRs), which play a pivotal role in modulating the plasticity of PPI networks, were found to be significantly enriched in cancer-specific PPIs in the human proteome [43][45]. Interestingly, a recent study found that protein hubs in cancer-specific PPIs tend to possess more distinct binding sites for various protein partners than non-cancer related proteins [44][46]. These findings indicate that cancer-specific hubs may have acquired unique structural features to coordinate diverse modules for maintaining the high plasticity and complexity of cancer networks. Of particular interest here, OGT and OGA are potential multi-interface hubs in PPIs, in agreement with their capability to accommodate remarkably diverse protein substrates and the fact that O-GlcNAcylation is often detected in the disordered regions of proteins [45][47]. While still far from a complete understanding of the protein recognition mechanisms of OGT/OGA, the structural features discussed below start to reveal the molecular basis underlying the selectivity and plasticity of their protein interactions, supporting that the O-GlcNAc cycling enzymes are essential regulators of the dynamic, scale-free PPI networks in cancer.3. Systematic Analyses of OGT/OGA Associated PPI Networks in Cancer
O-GlcNAc cycling enzymes (OGT and OGA) operate their functions by interactions with other biomolecules. The multiprotein complexes of OGT/OGA are of fundamental importance to decipher their roles in various biological processes. As previously reported that aberrant PPIs underlie the etiology of cancer, decoding the molecular connections of dysregulated OGT/OGA–protein networks in cancer will be important for therapeutic innovations [46][85]. To date, rapidly accumulating knowledge of O-GlcNAcylated proteins, and a few high-throughput studies of OGT interactions including the analyses using protein microarray in vitro [47][86] and the quantitative proteomics in mouse embryonic fibroblast (MEF) cells [48][87], have enabled the establishment of a massive compendium about the OGT interacting proteins (OGT-PIN) [49][88]. Less information about OGA binding partners has been disclosed; however, a potential high-level of overlap may exist between OGT– and OGA–substrate interactions. Despite these propitious findings, only a few systematic analyses of OGT/OGA-associated PPI networks have been reported in cancer models. The profiling of OGT/OGA-associated PPIs in cancer cells typically apply affinity purification or proximity biotinylation coupled with quantitative LC-MS/MS analysis (AP-MS or BioID-MS) [50][51][89,90]. For instance, the functions of mOGT in breast cancer cells have been investigated through its interactomes [52][91]. Compared to ncOGT, the relatively short TPR region (9 instead of 13.5 TPRs) and the unique mitochondrial localization imply that mOGT may form a PPI network different from ncOGT. This is in agreement with the distinct substrate profiles and cytotoxic effects of mOGT observed in mammalian cells [53][54][92,93]. Following endogenous ncOGT knockdown and HaloTag-mOGT affinity purification from mitochondrial fractions, more than 40 mitochondrial proteins have been identified as mOGT binding partners in at least two different breast cancer cell lines compared to HaloTag control [52][91]. These proteins participate in almost every aspect of mitochondrial functions, including mitochondrial transport, respiration, translation, fatty acid metabolism, apoptosis, and mtDNA processes. This finding is also in line with the observation in cervical cancer HeLa cells that mOGT contributes to mitochondrial structure and function, as well as cancer cell survival [55][94]. Surprisingly, a few nuclear proteins were also detected as mOGT binders. This implicates potentially distinct roles of different OGT isoforms in cancer cells. While these discoveries on mOGT–protein interactions are informative, further analyses will be needed to define cancer-specific PPIs of mOGT. Protein interactions with O-GlcNAc cycling enzymes consist of transient or weak interactions. The recently developed proximity biotinylation (BioID) technique is well-suited for this type of detection [51][90]. It was applied to investigate OGA-mediated oxidative stress response in osteosarcoma U2OS cells [56][95]. In this study, ectopic expression of OGA fused with biotin ligase mBirA can biotinylate proteins bound or in proximity to OGA. The changes of OGA–protein interactions in response to H2O2-induced oxidative stress were identified by LC-MS/MS detection of biotinylated proteins. As a result, dozens of OGA binding partners have been identified as significantly regulated, including fatty acid synthase (FAS), filamin-A (FLNA), heat shock cognate 70-kDa protein (HSC70), and OGT. Interestingly, biochemical analyses further revealed that the interaction with FAS suppressed OGA’s catalytic activity and modulated the stress adaptation of cancer cells. Using the AP-MS approach, another study identified OGA–protein interactions in HeLa cells [18], showing significant enrichment of cellular functions, such as RNA splicing, mRNA processing, cytoskeleton organization, intracellular transport, and mitosis (GO term analysis of the data from Table S1 in [18] using DAVID [57][58][96,97]). Intriguingly, many of these OGA PPI functions were absent in the OGA pHAT domain mutant (Y891F), except for RNA splicing and mRNA processing (GO term analysis of the data from Table S2 in [18] using DAVID), suggesting that the pHAT domain is indispensable for maintaining the integrity of OGA PPI networks. Notably, the same study also found that OGA was upregulated in many types of cancer and drove aerobic glycolysis and tumor growth by inhibiting pyruvate kinase M2 (PKM2). Further experiments suggested that the activity of PKM2 was dysregulated by OGA complex-associated acetylation and O-GlcNAcylation under cancer-related high glucose conditions. Overall, these studies have begun to uncover the abnormal PPIs of OGT/OGA in cancer models. With advances in proteomics and bioinformatics, the researchers envision that the systematic analyses of protein interactions with OGT/OGA (not restricted to O-GlcNAcylated proteins) will identify new, cancer-specific PPIs and help define the oncogenic properties of these O-GlcNAc cycling enzymes in cancer biology.4. Dysregulated Protein Functions by Rewired OGT/OGA Protein Networks in Cancer
PPIs are the frameworks for signal transmission in conducting cellular events. The broad-spectrum effect of O-GlcNAcylation suggest that OGT/OGA PPIs regulate the spatiotemporal communication of many biological processes. Analysis of all reported interacting partners of human OGT/OGA in the curated databases, OGT-PIN (high-stringency partners) [49][88] and PINA [59][43], demonstrated diverse molecular characters, including nucleotide binder, kinase/phosphatase, E3 ubiquitin ligase/deubiquitinase (DUB), and cytoskeleton (Figure 2a). Herein, abnormal OGT/OGA networks can affect proteins at multiple levels, including PTM, conformation, and association with other biomolecules, which consequently modulate the enzyme activity, protein stability and transportation, among others [7][9][7,9]. Below, the researchers highlight a few representative examples, in which the OGT/OGA–protein interactions have been validated by orthogonal methods, such as immunoprecipitation, to demonstrate the diverse molecular impacts of these PPIs on the malignant programming of cancer cells (Figure 2b). These studies illustrate that O-GlcNAc cycling enzymes can form divergent protein complexes with substrates and/or non-substrate partners and execute multifunctional roles in cancer. While most studies were focused on OGT, it is likely that OGA could apply similar mechanisms.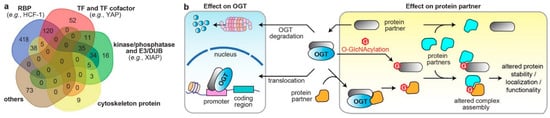
Figure 2. The diverse molecular impacts of protein interactions with O-GlcNAc cycling enzymes. (a) Classification of reported OGT/OGA binding partners in cancer. The information of OGT and OGA binding partners was from database OGT-PIN (high-stringency interaction proteins) and PINA, respectively. The binding partners were categorized using the databases AnimalTFDB 3.0 [60][98], KinMap [61][99], DEPOD [62][100], UbiBrowser 2.0 [63][101], UbiNet 2.0 [64][102], RBP2GO [65][103], and gene ontology (GO term: cytoskeleton) from DAVID [58][97]. The venn diagram was generated from http://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed on 15 October 2022). RBP, RNA binding protein; TF, transcription factor; E3, E3 ubiquitin ligase; DUB, deubiquitinase. (b) Different molecular mechanisms underlying the protein interactions with O-GlcNAc cycling enzymes in dysregulating protein functions in cancer cells (OGT is shown as an example).