Meiosis is the unique division of germ cells resulting in the recombination of the maternal and paternal genomes and the production of haploid gametes. In mammals, it begins during the fetal life in females and during puberty in males. In both cases, entering meiosis requires a timely switch from the mitotic to the meiotic cell cycle and the transition from a potential pluripotent status to meiotic differentiation. Revealing the molecular mechanisms underlying these interrelated processes represents the essence in understanding the beginning of meiosis. Meiosis facilitates diversity across individuals and acts as a fundamental driver of evolution. Major differences between sexes and among species complicate the understanding of how meiosis begins.
- meiosis
- PGC reprogramming
1. Introduction
2. Molecules and Conditions for Meiotic Beginning
2.1. Intrinsic Factors
The beginning of meiosis in germ cells requires the acquisition of a molecular status, critically including epigenomic and specific RNA-binding proteins, referable to what early researcher conceived as “intrinsic factors” (features not directly traceable to the action of specific external signaling) and a variety of local signals and conditions constituting the “extrinsic factors” (Figure 1).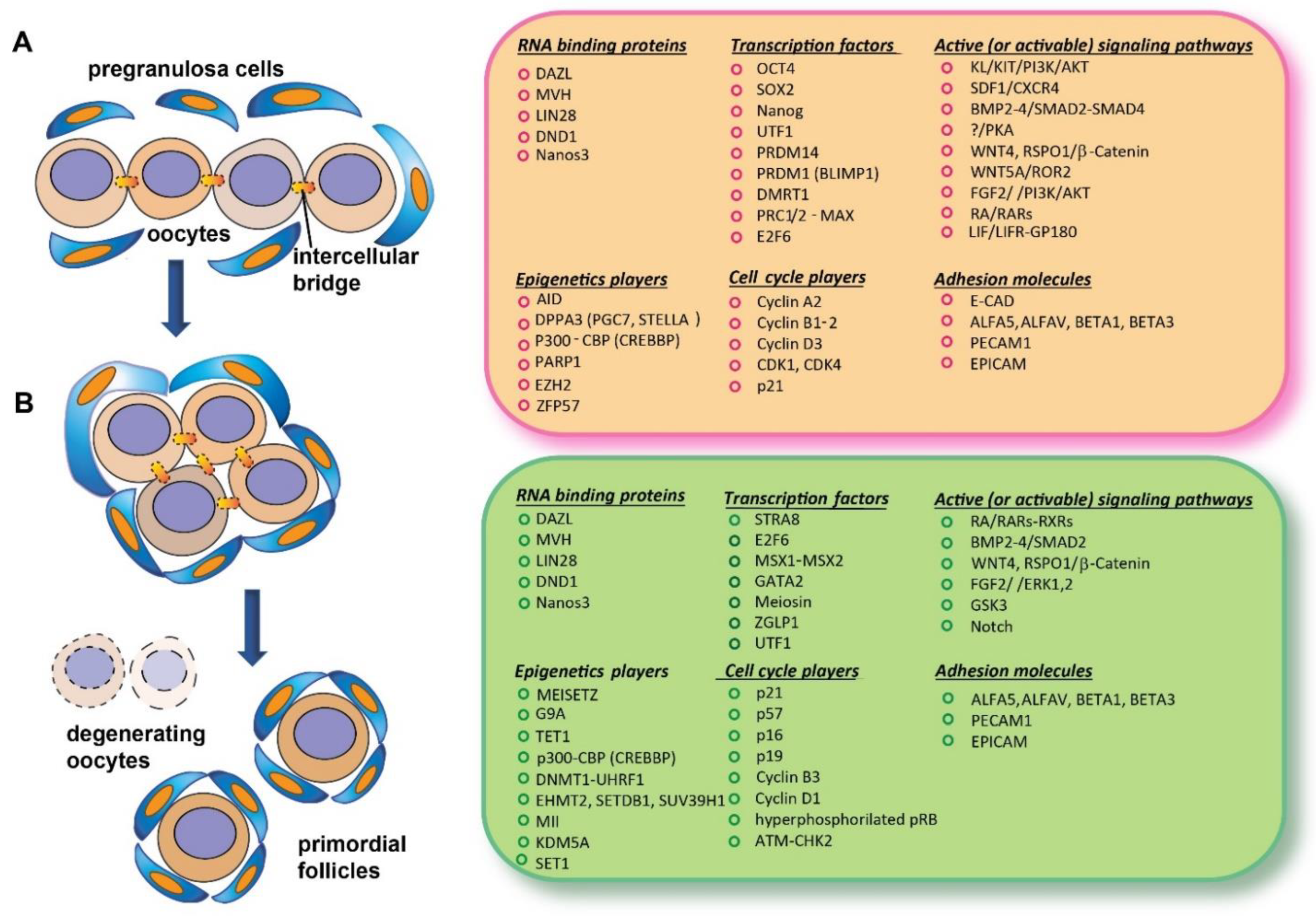
2.1.1. Genome-Wide Reprogramming in Premeiotic PGCs
2.1.2. DAZL and MVH
Substantial evidence supports the notion that in mice and probably in all mammalian species, RBP DAZL is critical for rendering female PGCs/oogonia competent to enter meiosis within the fetal ovary and for sex-specific differentiation events of oogenesis and spermatogenesis [80,81,82][35][36][37]. Despite this, the gametogenic functions of DAZL have not yet been fully characterized. DAZL was localized in both the nucleus and cytoplasm of fetal germ cells [83][38]. As reported in the previous section, its expression is epigenetically regulated by the DNA methylation of CpG islands present in its promoter region. Moreover, the fact that the first exon and intron of the gene are highly enriched in the active histone H3K4me3 mark and lack repressive H3K27me3 [84][39] suggests that promoter demethylation can rapidly lead to gene activation. The expression of the Dazl gene, however, might also depend on signals from the gonadal ridges, since in the absence of these, PGCs of both sexes were shown not to undergo gametogenesis and to retain their pre-gonadal characteristics [58][21]. Dazl and Mvh (see below) are not likely direct targets of RA, one of the major extrinsic factors triggering meiosis in mammalian germ cells, since they were not reported to possess Retinoic Acid Response Elements (RAREs) in mammals. Intriguingly, however, a functional Retinoic Acid Receptor beta (RARβ)-binding RARE sequence was found in the chicken Dazl gene core promoter region [85][40]. Deleted in azoospermia, the DAZ family consists of three members, DAZL, DAZ, and BOULE, which are exclusively expressed in pluripotent stem cells and germ cells [88][41]. As RBPs, these factors elicit their roles through modulating the translation and stabilization of specific mRNAs [89][42]. For example, DAZL, either directly, through 3′-UTR, or indirectly, through polyA-binding protein PABP, binds the mRNAs of germ-cell-specific genes, such as Mvh, Sycp1, Sycp3, Tex11, and Tex14, and facilitates the translation of their coded proteins [89,90][42][43]. Notably, in mouse embryos, depending on the genetic background, the ablation of Dazl causes a severe reduction in germ-cell numbers and the aberrant expression of markers of pluripotency in post-migratory PGCs. Moreover, these cells show the inability to differentiate along the oogenesis or spermatogenesis pathway and in female germ cells to enter or progress through meiosis [4,80,81][35][36][44]. By profiling gene expression in mouse fetal ovary mutants, Soh et al. recently reported that DAZL is required for the induction of nearly all 104 genes that they identified to be specifically expressed during meiotic prophase [91][45], but the way in which it controls such transcriptional program remains to be clarified. A study documented that GASZ, a protein with four ankyrin repeats encoded by an evolutionarily conserved gene expressed exclusively in germ cells, interacts with DAZL and synergistically stimulates PGCLCs to form mouse ESCs [92][46]. Evidence exists indicating that DAZL also plays a crucial role in human oogenesis [93][47]. In the human embryo, the percentage of germ cells in the fetal ovary highly expressing DAZL was found to be increased markedly from 28 to 48% from weeks 10 to 18 of development, while that of OCT4-positive cells was found to be decreased significantly. Notably, cells expressing a high level of DAZL almost always lacked OCT4 expression, indicating a mutually exclusive expression pattern of the pluripotency marker and DAZL [94][48]. Moreover, a combination of DAZL and BOULE could be utilized to induce human ESCs to exit the pluripotent state and enter meiosis in vitro [94][48]. The identification of RNA codifying genes involved in chromosome cohesion and DNA recombination as targets of human DAZL (SYCP3 and TEX19, for example) highlights the importance of this RBP also in early meiosis [95][49]. Similar to Dazl, Mvh (also known as Vasa or Ddx4) is expressed in germ cells around the time of PGC arrival at the gonadal ridges and requires the gonad environment for expression [96][50]. Although Mvh expression is independent of DAZL [58][21], the latter is able to bind Mvh RNA and modulate its translation [90][43]. Moreover, as reported above, Mvh expression is partly dependent on demethylation [42][5]. MVH is an ATP-dependent RNA helicase that often changes the secondary structures of RNA during processes such as alternative splicing and protein translation initiation [97][51]. MVH interacts with ribonucleic acids through its conserved DEAD (Asp-Glu-Ala-Asp)-RNA-binding motif and exerts multiple roles in the germline of various species [98][52]. For example, it appears to be able to regulate the proliferation and pluripotency of PGCs and meiosis in male germ cells; however, no specific functions of the protein in mammal oogenesis were found. Indeed, the loss of Mvh in the mouse affects the number and differentiation of male germ cells but apparently not oogenesis [99][53].2.2. Extrinsic Factors
Once a specific epigenetic status and the expression of DAZL RBP are established, PGCs become responsive to the action of local ovary/mesonephros factors and conditions that though various intracellular molecular cascades, promote or inhibit in the developing ovaries and testes, respectively, their entering into meiotic prophase I. Such extrinsic factors cause a mitotic G0 block in male PGCs/prospermatogonia, and a switch from the mitotic to meiotic cycle and the full activation of meiotic genes in female PGCs/oogonia (Figure 2).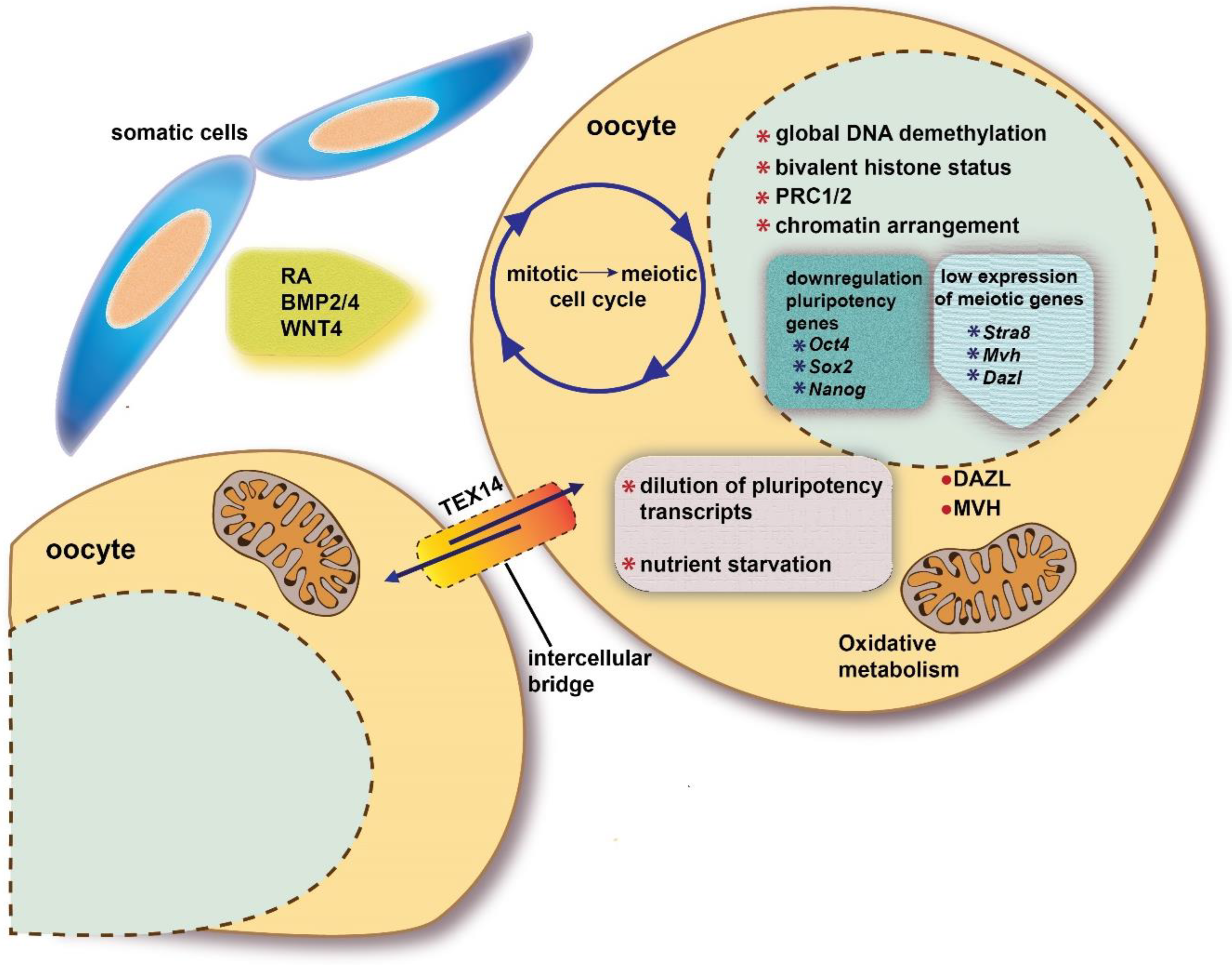
2.2.1. The Switch from Mitotic to Meiotic Cell Cycle
2.2.2. Retinoic Acid
2.2.3. STRA8
Stra8 was first identified in P19 teratocarcinoma cells as a RA-inducible gene and erroneously described as being restricted to male germ cells in fetal testes [143][80]. Gene knockout studies in mice clearly demonstrated that Stra8 is required for meiotic initiation and the meiotic progression of germ cells and that the ablation of the gene results in infertility in both sexes [6,7,8,9,10][73][81][82][83][84]. In mouse female embryos, Stra8-deficient PGCs do not initiate meiotic chromosome condensation, cohesion, and synapsis, or DNA double-strand breaks and recombination [6][81]. Additionally, different transcriptional (e.g., Pparg) and post-transcriptional regulators (e.g., Meioc and Ythdc2) involved in the control of meiotic prophase length (see above) are STRA8-regulated [91[45][65][66],111,112], thus indicating that other factors could participate in the STRA8 regulation of the meiotic program. In fact, how STRA8 exercises its transcriptional control is not completely clear. It is able to directly bind the genomic regulatory region close to the transcriptional start site (TSS) of the regulated genes [111,112][65][66] at a consensus motif (the CNCCTCAG sequence) that does not correspond to a E-Box sequence recognized by bHLH transcription regulators [155][85]. The same consensus region in the most meiotic-regulated genes is shared by Meiosin, a recently discovered STRA8 interactor in prospermatocytes [111][65]. This protein is also expressed in the mouse fetal ovary at the same time as STRA8, and ovarian germ cells fail to start meiosis when Meiosin is ablated as in Meiosin-/- spermatocytes. In addition, Meiosin, which possesses bHLH and HMG DNA-binding domains, similar to STRA8, controls a large transcriptional program, including meiotic and non-meiotic genes, most of which are also STRA8-regulated, thus indicating that Meiosin and STRA8 act in concert to amplify the network, ensuring the exact gene level indispensable for the beginning of meiosis at the right time. Do they conduct this by binding to open chromatin and recruiting RNA polymerase II, releasing it, or via other mechanisms? This is still an open question.2.3. Other Factors
2.3.1. Inhibitory and Cooperative Factors
The downregulation of the FGF9 and NODAL growth factors is considered as a prerequisite to make meiosis possible in the mouse embryonic ovary. FGF9, expressed in the somatic cells of both male and female undifferentiated GRs, remains highly expressed only in fetal testes, specifically in pre-Sertoli cells, mainly exerting a paracrine action on germ cells [29,159][86][87]. Conversely, NODAL, a member of the TGFβ superfamily, appears to be mainly expressed in PGCs and might act in an autocrine manner [137,160][88][89]. Intriguingly, in the human embryo, NODAL was found to be expressed in both female and male mitotic germ cells, whereas its receptor, ACVR1C, and target gene Pitx2 were specifically expressed in female meiotic and male mitotic-arrest germ cells [161][90]. Nonetheless, at least in the mouse, the absence of either FGF9 or NODAL can be considered permissive for meiosis onset rather than inductive. β-Catenin was also recently proposed to exert a negative action on the beginning of meiosis. Indeed, Le Rolle et al. [162][91] reported that β-Catenin was present in the nucleus of proliferating mouse PGCs physically associated with OCT4 and regulated chromatin accessibility to promoters and intergenic regions of several genes. The ablation of β-Catenin caused the exit from the mitotic cycle and the precocious expression of Dazl and meiotic genes including Stra8. In light of these and other results, the autscholars proposed that the activation of GSK3 and the upregulation of ZNRF3, an E3 ubiquitin-protein ligase that acts as a negative regulator of the WNT signaling pathway, in PGCs promote the inactivation of β-Catenin and contribute to the switch from mitosis to meiosis. Conversely, Activin A might cooperate with RA to promote meiosis. When Activin A was added to cultured 12.5 dpc mouse ovaries or delivered to 10.5 dpc litters via intraperitoneal injection, the expression of Stra8 and of meiotic genes in germ cells was increased. The activation mechanism was suggested to be indirect via the activation of SMAD3 in pre-granulosa cells and the downregulation of CYP26B1 [163][92].2.3.2. Nutrient and Metabolic Factors
Nutrient restrictions and changes in metabolisms were recently proposed to act in concert with RA stimulation to activate the meiotic program [167][93]. This is reminiscent of meiosis initiation in yeast, which mainly depends on nutrients and metabolism. Autophagy is crucial for reserving energy in response to cellular stress conditions such as nutrient and oxygen starvation. In both fission and budding yeast, meiotic entry fails if autophagy is deficient [170,171][94][95]. Autophagy might contribute to degrade major meiotic entry inhibitors, thus allowing the cell to enter meiosis [172][96]. However, in mammals, STRA8 represses autophagy by binding the promoter of NR1D1, which in turn leads to repressing its downstream target ULK1, an autophagy initiator, highlighting the requirement for the suppression of autophagy during meiosis initiation [173][97]. This means that although the role of nutrient deprivation in meiosis is potentially conserved, it is yet to be fully understood in mammals. It can be hypothesized that the suppression of autophagy via STRA8 might be important in maintaining meiotic DSBs during prophase I, as autophagy plays a role in DNA damage repair. The master regulator of autophagy, mTORC1, was found to be crucial during meiotic onset. In fact, in both yeast and female Drosophila, the reduction in TORC1 expression in response to nutrient starvation was needed for the mitotic–meiotic switch [174,175][98][99]. In mammals, the suppression of mTORC1 activators is required for male mitotic arrest in PGCs, which could prime male germ cells for meiotic entry [176][100].References
- Hajkova, P.; Ancelin, K.; Waldmann, T.; Lacoste, N.; Lange, U.C.; Cesari, F.; Lee, C.; Almouzni, G.; Schneider, R.; Surani, M.A. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 2008, 452, 877–881.
- Kafri, T.; Ariel, M.; Brandeis, M.; Shemer, R.; Urven, L.; McCarrey, J.; Cedar, H.; Razin, A. Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev. 1992, 6, 705–714.
- Leitch, H.G.; Tang, W.W.; Surani, M.A. Primordial germ-cell development and epigenetic reprogramming in mammals. Curr. Top. Dev. Biol. 2013, 104, 149–187.
- Seisenberger, S.; Andrews, S.; Krueger, F.; Arand, J.; Walter, J.; Santos, F.; Popp, C.; Thienpont, B.; Dean, W.; Reik, W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell 2012, 48, 849–862.
- Maatouk, D.M.; Kellam, L.D.; Mann, M.R.; Lei, H.; Li, E.; Bartolomei, M.S.; Resnick, J.L. DNA methylation is a primary mechanism for silencing postmigratory primordial germ cell genes in both germ cell and somatic cell lineages. Development 2006, 133, 3411–3418.
- Popp, C.; Dean, W.; Feng, S.; Cokus, S.J.; Andrews, S.; Pellegrini, M.; Jacobsen, S.E.; Reik, W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 2010, 463, 1101–1105.
- Ciccarone, F.; Klinger, F.G.; Catizone, A.; Calabrese, R.; Zampieri, M.; Bacalini, M.G.; De Felici, M.; Caiafa, P. Poly(ADP-ribosyl)ation acts in the DNA demethylation of mouse primordial germ cells also with DNA damage-independent roles. PLoS ONE 2012, 7, e46927.
- Hajkova, P.; Jeffries, S.J.; Lee, C.; Miller, N.; Jackson, S.P.; Surani, M.A. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science 2010, 329, 78–82.
- Kagiwada, S.; Kurimoto, K.; Hirota, T.; Yamaji, M.; Saitou, M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 2013, 32, 340–353.
- Tang, W.W.C.; Dietmann, S.; Irie, N.; Leitch, H.G.; Floros, V.I.; Bradshaw, C.R.; Hackett, J.A.; Chinnery, P.F.; Surani, M.A. A Unique Gene Regulatory Network Resets the Human Germline Epigenome for Development. Cell 2015, 161, 1453–1467.
- Kurimoto, K.; Yamaji, M.; Seki, Y.; Saitou, M. Specification of the germ cell lineage in mice: A process orchestrated by the PR-domain proteins, Blimp1 and Prdm14. Cell Cycle 2008, 7, 3514–3518.
- Hargan-Calvopina, J.; Taylor, S.; Cook, H.; Hu, Z.; Lee, S.A.; Yen, M.R.; Chiang, Y.S.; Chen, P.Y.; Clark, A.T. Stage-Specific Demethylation in Primordial Germ Cells Safeguards against Precocious Differentiation. Dev. Cell 2016, 39, 75–86.
- Matsui, Y.; Hayashi, K. Epigenetic regulation for the induction of meiosis. Cell. Mol. Life Sci CMLS 2007, 64, 257–262.
- Sachs, M.; Onodera, C.; Blaschke, K.; Ebata, K.T.; Song, J.S.; Ramalho-Santos, M. Bivalent chromatin marks developmental regulatory genes in the mouse embryonic germline in vivo. Cell Rep. 2013, 3, 1777–1784.
- De Felici, M. Nuclear reprogramming in mouse primordial germ cells: Epigenetic contribution. Stem Cells Int. 2011, 2011, 425863.
- Yokobayashi, S.; Liang, C.Y.; Kohler, H.; Nestorov, P.; Liu, Z.; Vidal, M.; van Lohuizen, M.; Roloff, T.C.; Peters, A.H. PRC1 coordinates timing of sexual differentiation of female primordial germ cells. Nature 2013, 495, 236–240.
- Fu, X.F.; Yang, F.; Cheng, S.F.; Feng, Y.N.; Li, L.; Dyce, P.W.; Shen, W.; Sun, X.F. The epigenetic modifications and the anterior to posterior characterization of meiotic entry during mouse oogenesis. Histochem. Cell Biol. 2017, 148, 61–72.
- Kawabata, Y.; Kamio, A.; Jincho, Y.; Sakashita, A.; Takashima, T.; Kobayashi, H.; Matsui, Y.; Kono, T. Sex-specific histone modifications in mouse fetal and neonatal germ cells. Epigenomics 2019, 11, 543–561.
- Mochizuki, K.; Tachibana, M.; Saitou, M.; Tokitake, Y.; Matsui, Y. Implication of DNA Demethylation and Bivalent Histone Modification for Selective Gene Regulation in Mouse Primordial Germ Cells. PLoS ONE 2012, 7, e46036.
- Hackett, J.A.; Sengupta, R.; Zylicz, J.J.; Murakami, K.; Lee, C.; Down, T.A.; Surani, M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 2013, 339, 448–452.
- Hu, Y.-C.; Nicholls, P.K.; Soh, Y.Q.S.; Daniele, J.R.; Junker, J.P.; van Oudenaarden, A.; Page, D.C. Licensing of Primordial Germ Cells for Gametogenesis Depends on Genital Ridge Signaling. PLOS Genet. 2015, 11, e1005019.
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935.
- Vincent, J.J.; Huang, Y.; Chen, P.Y.; Feng, S.; Calvopiña, J.H.; Nee, K.; Lee, S.A.; Le, T.; Yoon, A.J.; Faull, K.; et al. Stage-specific roles for tet1 and tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell 2013, 12, 470–478.
- Yamaguchi, S.; Hong, K.; Liu, R.; Shen, L.; Inoue, A.; Diep, D.; Zhang, K.; Zhang, Y. Tet1 controls meiosis by regulating meiotic gene expression. Nature 2012, 492, 443–447.
- Hill, P.W.S.; Leitch, H.G.; Requena, C.E.; Sun, Z.; Amouroux, R.; Roman-Trufero, M.; Borkowska, M.; Terragni, J.; Vaisvila, R.; Linnett, S.; et al. Epigenetic reprogramming enables the transition from primordial germ cell to gonocyte. Nature 2018, 555, 392–396.
- Welling, M.; Chen, H.-H.; Muñoz, J.; Musheev, M.U.; Kester, L.; Junker, J.P.; Mischerikow, N.; Arbab, M.; Kuijk, E.; Silberstein, L.; et al. DAZL regulates Tet1 translation in murine embryonic stem cells. EMBO Rep. 2015, 16, 791–802.
- Ito, T.; Osada, A.; Ohta, M.; Yokota, K.; Nishiyama, A.; Niikura, Y.; Tamura, T.; Sekita, Y.; Kimura, T. SWI/SNF chromatin remodeling complex is required for initiation of sex-dependent differentiation in mouse germline. Sci. Rep. 2021, 11, 24074.
- Lawson, K.A.; Dunn, N.R.; Roelen, B.A.; Zeinstra, L.M.; Davis, A.M.; Wright, C.V.; Korving, J.P.; Hogan, B.L. Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 1999, 13, 424–436.
- Ohinata, Y.; Ohta, H.; Shigeta, M.; Yamanaka, K.; Wakayama, T.; Saitou, M. A signaling principle for the specification of the germ cell lineage in mice. Cell 2009, 137, 571–584.
- Saitou, M.; Barton, S.C.; Surani, M.A. A molecular programme for the specification of germ cell fate in mice. Nature 2002, 418, 293–300.
- Ohinata, Y.; Payer, B.; O’Carroll, D.; Ancelin, K.; Ono, Y.; Sano, M.; Barton, S.C.; Obukhanych, T.; Nussenzweig, M.; Tarakhovsky, A.; et al. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature 2005, 436, 207–213.
- Yamaji, M.; Seki, Y.; Kurimoto, K.; Yabuta, Y.; Yuasa, M.; Shigeta, M.; Yamanaka, K.; Ohinata, Y.; Saitou, M. Critical function of Prdm14 for the establishment of the germ cell lineage in mice. Nat. Genet. 2008, 40, 1016–1022.
- De Felici, M.; Pesce, M. Growth factors in mouse primordial germ cell migration and proliferation. Prog. Growth Factor Res. 1994, 5, 135–143.
- De Felici, M. Primordial germ cell biology at the beginning of the XXI century. Int. J. Dev. Biol. 2009, 53, 891–894.
- Ruggiu, M.; Speed, R.; Taggart, M.; McKay, S.J.; Kilanowski, F.; Saunders, P.; Dorin, J.; Cooke, H.J. The mouse Dazla gene encodes a cytoplasmic protein essential for gametogenesis. Nature 1997, 389, 73–77.
- Schrans-Stassen, B.H.; Saunders, P.T.; Cooke, H.J.; de Rooij, D.G. Nature of the spermatogenic arrest in Dazl −/− mice. Biol. Reprod. 2001, 65, 771–776.
- Gill, M.E.; Hu, Y.C.; Lin, Y.; Page, D.C. Licensing of gametogenesis, dependent on RNA binding protein DAZL, as a gateway to sexual differentiation of fetal germ cells. Proc. Natl. Acad. Sci. USA 2011, 108, 7443–7448.
- Reijo, R.A.; Dorfman, D.M.; Slee, R.; Renshaw, A.A.; Loughlin, K.R.; Cooke, H.; Page, D.C. DAZ family proteins exist throughout male germ cell development and transit from nucleus to cytoplasm at meiosis in humans and mice. Biol. Reprod. 2000, 63, 1490–1496.
- Sekinaka, T.; Hayashi, Y.; Noce, T.; Niwa, H.; Matsui, Y. Selective de-repression of germ cell-specific genes in mouse embryonic fibroblasts in a permissive epigenetic environment. Sci. Rep. 2016, 6, 32932.
- Zhang, L.; Zhu, R.; Zuo, Q.; Li, D.; Lian, C.; Tang, B.; Xiao, T.; Zhang, Y.; Li, B. Activity analysis and preliminary inducer screening of the chicken DAZL gene promoter. Int. J. Mol. Sci. 2015, 16, 6595–6605.
- Fu, X.-F.; Cheng, S.-F.; Wang, L.-Q.; Yin, S.; De Felici, M.; Shen, W. DAZ Family Proteins, Key Players for Germ Cell Development. Int. J. Biol. Sci. 2015, 11, 1226–1235.
- Collier, B.; Gorgoni, B.; Loveridge, C.; Cooke, H.J.; Gray, N.K. The DAZL family proteins are PABP-binding proteins that regulate translation in germ cells. EMBO J. 2005, 24, 2656–2666.
- Reynolds, N.; Collier, B.; Maratou, K.; Bingham, V.; Speed, R.M.; Taggart, M.; Semple, C.A.; Gray, N.K.; Cooke, H.J. Dazl binds in vivo to specific transcripts and can regulate the pre-meiotic translation of Mvh in germ cells. Hum. Mol. Genet. 2005, 14, 3899–3909.
- Lin, Y.; Gill, M.E.; Koubova, J.; Page, D.C. Germ cell-intrinsic and -extrinsic factors govern meiotic initiation in mouse embryos. Science 2008, 322, 1685–1687.
- Soh, Y.Q.S.; Junker, J.P.; Gill, M.E.; Mueller, J.L.; van Oudenaarden, A.; Page, D.C. A Gene Regulatory Program for Meiotic Prophase in the Fetal Ovary. PLOS Genet. 2015, 11, e1005531.
- Wang, Q.; Liu, X.; Tang, N.; Archambeault, D.R.; Li, J.; Song, H.; Tang, C.; He, B.; Matzuk, M.M.; Wang, Y. GASZ promotes germ cell derivation from embryonic stem cells. Stem Cell Res. 2013, 11, 845–860.
- Rosario, R.; Adams, I.R.; Anderson, R.A. Is there a role for DAZL in human female fertility? Mol. Hum. Reprod. 2016, 22, 377–383.
- Jung, D.; Xiong, J.; Ye, M.; Qin, X.; Li, L.; Cheng, S.; Luo, M.; Peng, J.; Dong, J.; Tang, F.; et al. In vitro differentiation of human embryonic stem cells into ovarian follicle-like cells. Nat. Commun. 2017, 8, 15680.
- Rosario, R.; Smith, R.W.; Adams, I.R.; Anderson, R.A. RNA immunoprecipitation identifies novel targets of DAZL in human foetal ovary. Mol. Hum. Reprod. 2017, 23, 177–186.
- Toyooka, Y.; Tsunekawa, N.; Takahashi, Y.; Matsui, Y.; Satoh, M.; Noce, T. Expression and intracellular localization of mouse Vasa-homologue protein during germ cell development. Mech. Dev. 2000, 93, 139–149.
- Lasko, P. The DEAD-box helicase Vasa: Evidence for a multiplicity of functions in RNA processes and developmental biology. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2013, 1829, 810–816.
- Xu, C.; Cao, Y.; Bao, J. Building RNA-protein germ granules: Insights from the multifaceted functions of DEAD-box helicase Vasa/Ddx4 in germline development. Cell. Mol. Life Sci. 2021, 79, 4.
- Tanaka, S.S.; Toyooka, Y.; Akasu, R.; Katoh-Fukui, Y.; Nakahara, Y.; Suzuki, R.; Yokoyama, M.; Noce, T. The mouse homolog of Drosophila Vasa is required for the development of male germ cells. Genes Dev. 2000, 14, 841–853.
- De Miguel, M.P.; Alcaina, Y.; de la Maza, D.S. Primordial germ cell reprogramming. In Germ Cell; IntechOpen: London, UK, 2018; pp. 43–62.
- Donovan, P.J.; de Miguel, M.P. Turning germ cells into stem cells. Curr. Opin. Genet. Dev. 2003, 13, 463–471.
- Overeem, A.W.; Chang, Y.W.; Spruit, J.; Roelse, C.M.; Chuva De Sousa Lopes, S.M. Ligand-Receptor Interactions Elucidate Sex-Specific Pathways in the Trajectory from Primordial Germ Cells to Gonia During Human Development. Front. Cell Dev. Biol. 2021, 9, 661243.
- Western, P.S.; Miles, D.C.; van den Bergen, J.A.; Burton, M.; Sinclair, A.H. Dynamic regulation of mitotic arrest in fetal male germ cells. Stem Cells 2008, 26, 339–347.
- Spiller, C.; Wilhelm, D.; Koopman, P. Cell cycle analysis of fetal germ cells during sex differentiation in mice. Biol. Cell 2009, 101, 587–598.
- Watanabe, Y.; Yokobayashi, S.; Yamamoto, M.; Nurse, P. Pre-meiotic S phase is linked to reductional chromosome segregation and recombination. Nature 2001, 409, 359–363.
- Forsburg, S.L.; Hodson, J.A. Mitotic replication initiation proteins are not required for pre-meiotic S phase. Nat. Genet. 2000, 25, 263–268.
- Murakami, H.; Nurse, P. Regulation of premeiotic S phase and recombination-related double-strand DNA breaks during meiosis in fission yeast. Nat. Genet. 2001, 28, 290–293.
- Callan, H.G. Replication of DNA in the chromosomes of eukaryotes. Proc. R. Soc. London. Ser. B Biol. Sci. 1972, 181, 19–41.
- Lee, B.; Amon, A. Meiosis: How to create a specialized cell cycle. Curr. Opin. Cell Biol. 2001, 13, 770–777.
- Williamson, D.H.; Johnston, L.H.; Fennell, D.J.; Simchen, G. The timing of the S phase and other nuclear events in yeast meiosis. Exp. Cell Res. 1983, 145, 209–217.
- Ishiguro, K.I.; Matsuura, K.; Tani, N.; Takeda, N.; Usuki, S.; Yamane, M.; Sugimoto, M.; Fujimura, S.; Hosokawa, M.; Chuma, S.; et al. MEIOSIN Directs the Switch from Mitosis to Meiosis in Mammalian Germ Cells. Dev. Cell 2020, 52, 429–445.e10.
- Kojima, M.L.; de Rooij, D.G.; Page, D.C. Amplification of a broad transcriptional program by a common factor triggers the meiotic cell cycle in mice. eLife 2019, 8, e43738.
- Kumar, S.; Chatzi, C.; Brade, T.; Cunningham, T.J.; Zhao, X.; Duester, G. Sex-specific timing of meiotic initiation is regulated by Cyp26b1 independent of retinoic acid signalling. Nat. Commun. 2011, 2, 151.
- Vernet, N.; Condrea, D.; Mayere, C.; Féret, B.; Klopfenstein, M.; Magnant, W.; Alunni, V.; Teletin, M.; Souali-Crespo, S.; Nef, S.; et al. Meiosis occurs normally in the fetal ovary of mice lacking all retinoic acid receptors. Sci. Adv. 2020, 6, eaaz1139.
- Chassot, A.A.; Le Rolle, M.; Jolivet, G.; Stevant, I.; Guigonis, J.M.; Da Silva, F.; Nef, S.; Pailhoux, E.; Schedl, A.; Ghyselinck, N.B.; et al. Retinoic acid synthesis by ALDH1A proteins is dispensable for meiosis initiation in the mouse fetal ovary. Sci. Adv. 2020, 6, eaaz1261.
- Bellutti, L.; Abby, E.; Tourpin, S.; Messiaen, S.; Moison, D.; Trautmann, E.; Guerquin, M.-J.; Rouiller-Fabre, V.; Habert, R.; Livera, G. Divergent Roles of CYP26B1 and Endogenous Retinoic Acid in Mouse Fetal Gonads. Biomolecules 2019, 9, 536.
- Endo, T.; Mikedis, M.M.; Nicholls, P.K.; Page, D.C.; de Rooij, D.G. Retinoic Acid and Germ Cell Development in the Ovary and Testis. Biomolecules 2019, 9, 775.
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931.
- Bowles, J.; Knight, D.; Smith, C.; Wilhelm, D.; Richman, J.; Mamiya, S.; Yashiro, K.; Chawengsaksophak, K.; Wilson, M.J.; Rossant, J.; et al. Retinoid signaling determines germ cell fate in mice. Science 2006, 312, 596–600.
- Mu, X.; Wen, J.; Guo, M.; Wang, J.; Li, G.; Wang, Z.; Wang, Y.; Teng, Z.; Cui, Y.; Xia, G. Retinoic acid derived from the fetal ovary initiates meiosis in mouse germ cells. J. Cell. Physiol. 2013, 228, 627–639.
- Bowles, J.; Feng, C.W.; Miles, K.; Ineson, J.; Spiller, C.; Koopman, P. ALDH1A1 provides a source of meiosis-inducing retinoic acid in mouse fetal ovaries. Nat. Commun. 2016, 7, 10845.
- Bullejos, M.; Koopman, P. Germ cells enter meiosis in a rostro-caudal wave during development of the mouse ovary. Mol. Reprod. Dev. 2004, 68, 422–428.
- Menke, D.B.; Koubova, J.; Page, D.C. Sexual differentiation of germ cells in XX mouse gonads occurs in an anterior-to-posterior wave. Dev. Biol. 2003, 262, 303–312.
- Giuili, G.; Tomljenovic, A.; Labrecque, N.; Oulad-Abdelghani, M.; Rassoulzadegan, M.; Cuzin, F. Murine spermatogonial stem cells: Targeted transgene expression and purification in an active state. EMBO Rep. 2002, 3, 753–759.
- Feng, C.-W.; Burnet, G.; Spiller, C.M.; Cheung, F.K.M.; Chawengsaksophak, K.; Koopman, P.; Bowles, J. Identification of regulatory elements required for Stra8 expression in fetal ovarian germ cells of the mouse. Development 2021, 148, dev194977.
- Oulad-Abdelghani, M.; Bouillet, P.; Décimo, D.; Gansmuller, A.; Heyberger, S.; Dollé, P.; Bronner, S.; Lutz, Y.; Chambon, P. Characterization of a premeiotic germ cell-specific cytoplasmic protein encoded by Stra8, a novel retinoic acid-responsive gene. J. Cell Biol. 1996, 135, 469–477.
- Baltus, A.E.; Menke, D.B.; Hu, Y.-C.; Goodheart, M.L.; Carpenter, A.E.; de Rooij, D.G.; Page, D.C. In germ cells of mouse embryonic ovaries, the decision to enter meiosis precedes premeiotic DNA replication. Nat. Genet. 2006, 38, 1430–1434.
- Koubova, J.; Menke, D.B.; Zhou, Q.; Capel, B.; Griswold, M.D.; Page, D.C. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 2474–2479.
- Anderson, E.L.; Baltus, A.E.; Roepers-Gajadien, H.L.; Hassold, T.J.; de Rooij, D.G.; van Pelt, A.M.M.; Page, D.C. Stra8 and its inducer, retinoic acid, regulate meiotic initiation in both spermatogenesis and oogenesis in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 14976–14980.
- Mark, M.; Jacobs, H.; Oulad-Abdelghani, M.; Dennefeld, C.; Féret, B.; Vernet, N.; Codreanu, C.A.; Chambon, P.; Ghyselinck, N.B. STRA8-deficient spermatocytes initiate, but fail to complete, meiosis and undergo premature chromosome condensation. J. Cell Sci. 2008, 121, 3233–3242.
- Massari, M.E.; Murre, C. Helix-loop-helix proteins: Regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 2000, 20, 429–440.
- Bowles, J.; Feng, C.W.; Spiller, C.; Davidson, T.L.; Jackson, A.; Koopman, P. FGF9 suppresses meiosis and promotes male germ cell fate in mice. Dev. Cell 2010, 19, 440–449.
- Barrios, F.; Filipponi, D.; Pellegrini, M.; Paronetto, M.P.; Di Siena, S.; Geremia, R.; Rossi, P.; De Felici, M.; Jannini, E.A.; Dolci, S. Opposing effects of retinoic acid and FGF9 on Nanos2 expression and meiotic entry of mouse germ cells. J. Cell Sci. 2010, 123, 871–880.
- Feng, Y.M.; Liang, G.J.; Pan, B.; Qin, X.S.; Zhang, X.F.; Chen, C.L.; Li, L.; Cheng, S.F.; De Felici, M.; Shen, W. Notch pathway regulates female germ cell meiosis progression and early oogenesis events in fetal mouse. Cell Cycle 2014, 13, 782–791.
- Souquet, B.; Tourpin, S.; Messiaen, S.; Moison, D.; Habert, R.; Livera, G. Nodal Signaling Regulates the Entry into Meiosis in Fetal Germ Cells. Endocrinology 2012, 153, 2466–2473.
- De Bellis, M.; Carbonara, R.; Roussel, J.; Farinato, A.; Massari, A.; Pierno, S.; Muraglia, M.; Corbo, F.; Franchini, C.; Carratu, M.R.; et al. Increased sodium channel use-dependent inhibition by a new potent analogue of tocainide greatly enhances in vivo antimyotonic activity. Neuropharmacology 2017, 113, 206–216.
- Le Rolle, M.; Massa, F.; Siggers, P.; Turchi, L.; Loubat, A.; Koo, B.K.; Clevers, H.; Greenfield, A.; Schedl, A.; Chaboissier, M.C.; et al. Arrest of WNT/β-catenin signaling enables the transition from pluripotent to differentiated germ cells in mouse ovaries. Proc. Natl. Acad. Sci. USA 2021, 118, e2023376118.
- Liang, G.J.; Zhang, X.F.; Wang, J.J.; Sun, Y.C.; Sun, X.F.; Cheng, S.F.; Li, L.; De Felici, M.; Shen, W. Activin A accelerates the progression of fetal oocytes throughout meiosis and early oogenesis in the mouse. Stem Cells Dev. 2015, 24, 2455–2465.
- Zhang, X.; Gunewardena, S.; Wang, N. Nutrient restriction synergizes with retinoic acid to induce mammalian meiotic initiation in vitro. Nat. Commun. 2021, 12, 1758.
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174.
- Matsuhara, H.; Yamamoto, A. Autophagy is required for efficient meiosis progression and proper meiotic chromosome segregation in fission yeast. Genes Cells Devoted Mol. Cell. Mech. 2016, 21, 65–87.
- Wen, F.P.; Guo, Y.S.; Hu, Y.; Liu, W.X.; Wang, Q.; Wang, Y.T.; Yu, H.Y.; Tang, C.M.; Yang, J.; Zhou, T.; et al. Distinct temporal requirements for autophagy and the proteasome in yeast meiosis. Autophagy 2016, 12, 671–688.
- Ferder, I.C.; Fung, L.; Ohguchi, Y.; Zhang, X.; Lassen, K.G.; Capen, D.; Brown, D.; Xavier, R.J.; Wang, N. Meiotic gatekeeper STRA8 suppresses autophagy by repressing Nr1d1 expression during spermatogenesis in mice. PLoS Genet. 2019, 15, e1008084.
- Zheng, X.-F.; Schreiber, S.L. Target of rapamycin proteins and their kinase activities are required for meiosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3070–3075.
- Wei, Y.; Reveal, B.; Reich, J.; Laursen, W.J.; Senger, S.; Akbar, T.; Iida-Jones, T.; Cai, W.; Jarnik, M.; Lilly, M.A. TORC1 regulators Iml1/GATOR1 and GATOR2 control meiotic entry and oocyte development in Drosophila. Proc. Natl. Acad. Sci. USA 2014, 111, E5670–E5677.
- Shimada, R.; Koike, H.; Hirano, T.; Saga, Y. NANOS2 suppresses the cell cycle by repressing mTORC1 activators in embryonic male germ cells. bioRxiv 2020.