Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Helene Benedetti.
Neurofibromin is a large and multifunctional protein encoded by the tumor suppressor gene NF1, mutations of which cause the tumor predisposition syndrome neurofibromatosis type 1 (NF1). Neurofibromatosis type 1 is an autosomal dominant disorder caused by inherited or de novo germline mutations in the NF1 tumor suppressor gene. It is the most common tumor-predisposing disease in humans. It affects approximately one in 3000 live births and patients present widely heterogeneous clinical manifestations, even within the same family.
- Neurofibromin
- structure
- function
- localization
- interactions
1. Ras-GAP Activity
Neurofibromin is a GTPase-activating protein of Ras (Ras-GAP). It downregulates the Ras signaling pathway by promoting the hydrolysis of the active form of Ras (GTP-bound Ras) to an inactive form of Ras (GDP-bound Ras) by increasing the intrinsic GTPase activity of Ras [20,21,22][1][2][3]. Ras was shown to be constitutively active in malignant tumor cell lines derived from NF1 patients, even though Ras and p120GAP were functionally wildtype, suggesting that neurofibromin is the main negative regulator of Ras in the tested tissues [82,83][4][5]. This may be explained by the fact that neurofibromin binds more efficiently to Ras than p120GAP [84][6].
1.1. Role in Cell Growth
GRD expression normalizes active Ras levels and restores normal growth of NF1-deficient cell lines derived from NF1 patients [83,85][5][7]. A reduction (NF1+/−) or loss (NF1−/−) of neurofibromin expression in neural stem cells (NSCs) confers survival and a proliferative advantage as a consequence of hyperactivation of the Ras signaling pathway that was rescued by GRD expression [86][8].
The duration of Ras signaling is critical for signaling decisions [87][9]. Stimulation by different growth factors can induce different durations of Ras activation and lead to different cell responses [87][9]. Cichowski et al. (2003) [88][10] showed that neurofibromin regulates the amplitude and duration of Ras signaling pathway activation in growth-factor responses. Indeed, NF1-deficient mouse embryonic fibroblasts (MEFs) have been shown to be more sensitive to growth factors than wildtype MEFs. They only require non-mitogenic levels of growth factors for maximal Ras activation and proliferation [88][10]. Consistent with this observation, NF1-deficient (NF1−/−) hematopoietic cells show a hyperactive Ras signaling pathway and a high number of colonies in cultures supplemented with low concentrations of stimulating factors [89][11]. Henning et al. (2016) [90][12] described a feedback mechanism leading to stimulation of neurofibromin GAP activity to restrict the duration of growth factor-mediated Ras activation. In this study, knockdown of neurofibromin prolonged Ras-GTP accumulation in cells stimulated by epidermal growth factor (EGF) [90][12].
The Ras isoform preferentially inhibited by neurofibromin may differ from one cell type to another. In astrocytes, neurofibromin loss results in selective hyperactivation of K-Ras. Activation of K-Ras, but not H-Ras, is the cause of the proliferative advantage in NF1−/− astrocytes [91][13]. This study provided evidence that K-Ras is the primary target of neurofibromin Ras-GAP activity and growth control in astrocytes. The authors found these observations to have important pharmacological and therapeutic implications. They suggested that molecular therapies targeting K-Ras may be the most appropriate choice in the treatment of NF1-associated tumors, in which K-Ras is specifically hyperactivated. Knowing that all Ras isoforms are farnesylated by farnesyltransferases, and that K-Ras is the only geranylgeranylated isoform, the use of geranylgeranyl transferase inhibitors alone or in combination with farnesyltransferase inhibitors (FTI) was suggested [91][13]. Unfortunately, a phase II trial using tipifarnib, a farnesyltransferase inhibitor, demonstrated no influence on time to progression of plexiform neurofibroma [92][14].
1.2. Role in Learning
Ras inhibition by neurofibromin has been shown to be involved in learning. Costa et al. (2002) [93][15] showed that learning deficits and impaired long-term potentiation LTP (LTP is a synaptic plasticity mechanism involved in learning and memory), seen in NF1+/− mice, were caused by Ras hyperactivation.
Genetic or pharmacological manipulation that decrease Ras/MAPK activity rescued these phenotypes, suggesting that Ras/MAPK inhibition could be a therapeutic strategy against learning deficits in NF1 patients [93,94,95,96][15][16][17][18]. Along this line, different clinical trials were performed on cognitive outcomes of NF1 children using statins medications (lovastatin and simvastatin, which inhibit Ras farnesylation) with no improvement in visuospatial learning or attention [97,98,99][19][20][21].
A further study in this context shed light on the molecular mechanisms involved in the learning disabilities in NF1 mouse models with heterozygous Cre-mediated deletion of NF1 (resulting in hyperactivation of the ERK signaling pathway) in different key cell types of the brain (astrocytes, pyramidal neurons, excitatory and inhibitory neurons, GABAergic neurons) [100][22]. An increase in gamma-aminobutyric acid (GABA) release was shown to take place in inhibitory neurons of the hippocampus, resulting from the ERK-dependent phosphorylation of synapsin I. Such an increase in GABA levels in the hippocampus affected hippocampal synaptic plasticity, LTP, and learning. Indeed, a GABA antagonist rescued learning deficits associated with NF1 deletion in inhibitory neurons [100][22] (Figure 81).
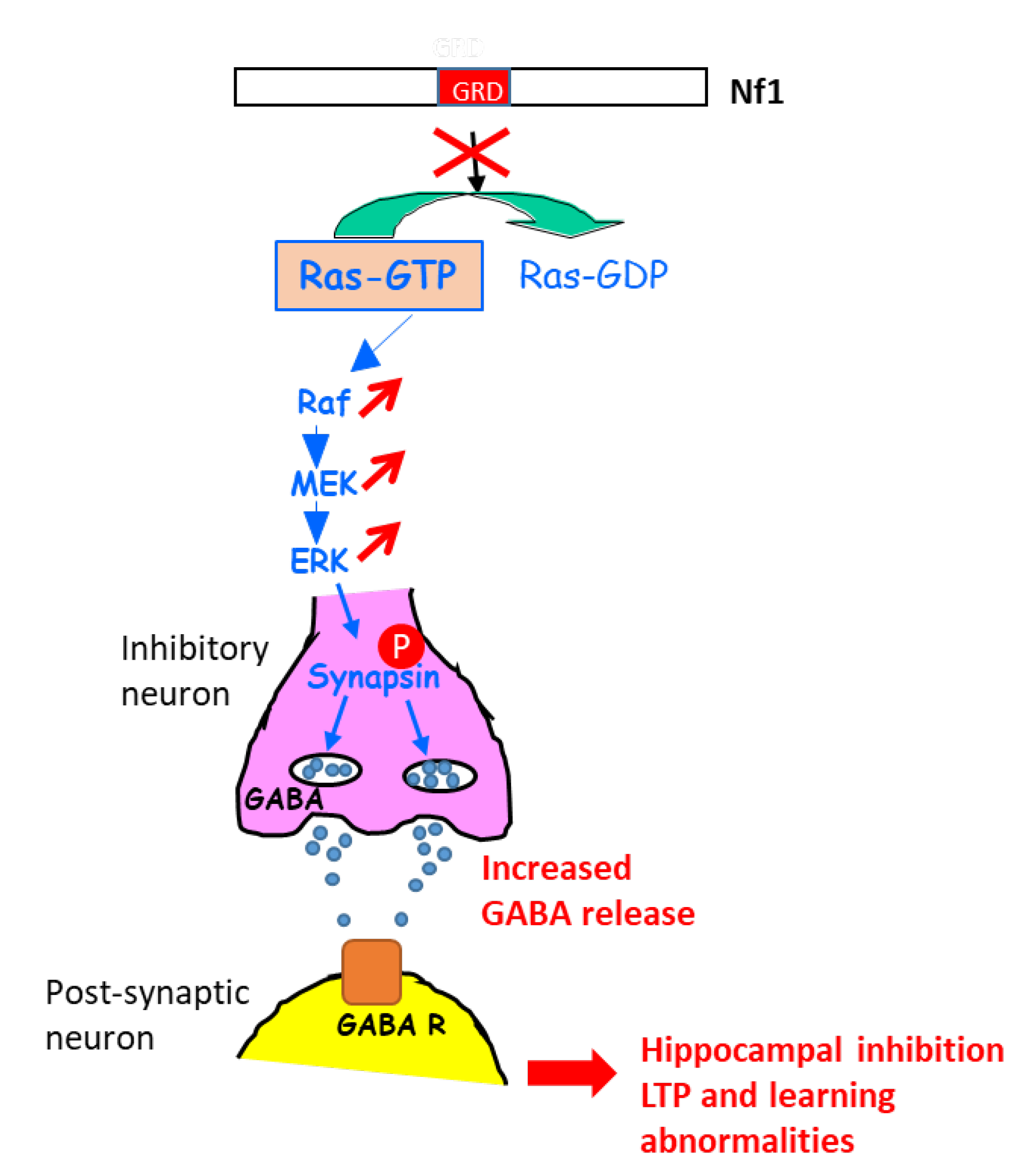
Figure 81. Schematic representation explaining the increase in GABA secretion upon the activation of ERK in NF1-deficient inhibitory neurons. GABA R: GABA receptor.
Oliveira and Yasuda (2014) [87][9] also reported that neurofibromin regulates synaptic plasticity via its Ras-GAP activity. In this study, the authors showed that neurofibromin is the major Ras inactivator in dendritic spines of pyramidal neurons of the hippocampus and that reduction of Ras inactivation in neurons expressing low levels of neurofibromin leads to impaired structural plasticity and spine loss. The observed phenotypes were rescued by the expression of the neurofibromin-GRD [87][9].
2. cAMP Regulation
In most mammalian cells, the cAMP-dependent protein kinase A pathway promotes growth arrest and cell differentiation [101][23]. In addition, cAMP disruption in NF1 mouse models has been shown to be sufficient to promote glioma formation, demonstrating the role of cAMP in the formation of certain tumors [102][24].
2.1. Neurofibromin is a Positive Regulator of cAMP Levels in Various Cell Types in Both a Ras-Dependent and Ras-Independent Manner
In Drosophila, neurofibromin is required to promote G protein-mediated adenylyl cyclase activation. Neurofibromin-null mutant flies have low cAMP levels [103][25]. They are characterized by small size and learning defects that are rescued by the expression of constitutively active PKA, the downstream effector of cAMP. Pharmacological enhancement of cAMP signaling also restores learning defects in NF1-deficient zebrafish [104][26].
However, in Drosophila, it is clear that these phenotypes are not rescued by attenuating Ras activity [105,106,107][27][28][29].
Similarly, GRD expression in NF1-deficient murine neocortical astrocytes only partially restores neurofibromin-mediated cAMP production in response to PACAP (pituitary adenylate cyclase activating peptide), suggesting that one or more other domains outside of the GRD are involved in Ras-independent neurofibromin regulation of cAMP levels [108][30] into these cells.
Furthermore, elevating cAMP levels with the adenylate cyclase (AC) activator forskolin or treatment with the phosphodiesterase 4 inhibitor rolipram, but not by MEK or PI3K inhibition, have been shown to reverse the decrease in neurite length, growth cone area, and the increase of apoptosis caused by impaired neurofibromin-mediated cAMP production in NF1-deficient mouse embryonic brain [109][31]. The same results have been obtained in NF1-heterozygous (NF1+/−) CNS, hippocampal, and retinal ganglion cell (RGC) neurons [110,111][32][33].
2.2. Detailed Molecular Mechanisms of Neurofibromin-Mediated cAMP Regulation
Further studies in Drosophila brain have shown that neurofibromin regulates cAMP by activating two distinct adenylate cyclase (AC) pathways. The first is a growth factor-stimulated neurofibromin/Ras-dependent AC pathway downstream of tyrosine kinase growth factor receptors (EGF, transforming growth factor TGF, etc.). This pathway requires Ras-GAP activity of the neurofibromin-GRD domain and Ras to stimulate AC activity, independently of Gαs. The second is a neurofibromin/Gαs-dependent pathway that is stimulated by neurotransmitters, such as serotonin and histamine. This pathway requires the CTD of neurofibromin [112][34]. The existence of this Ras-independent pathway is supported by a recent study showing that, by interacting with the serotonin receptor 5-HT6R via its PH domain, neurofibromin promotes 5-HT6R constitutive activation of the Gαs/AC pathway in striatal neurons. Both NF1 silencing and NF1 patient mutations within the PH domain that disrupt the neurofibromin/5-HT6R interaction inhibit constitutive receptor activity on the Gαs/AC pathway and reduce basal cAMP levels [113][35] (Figure 92).
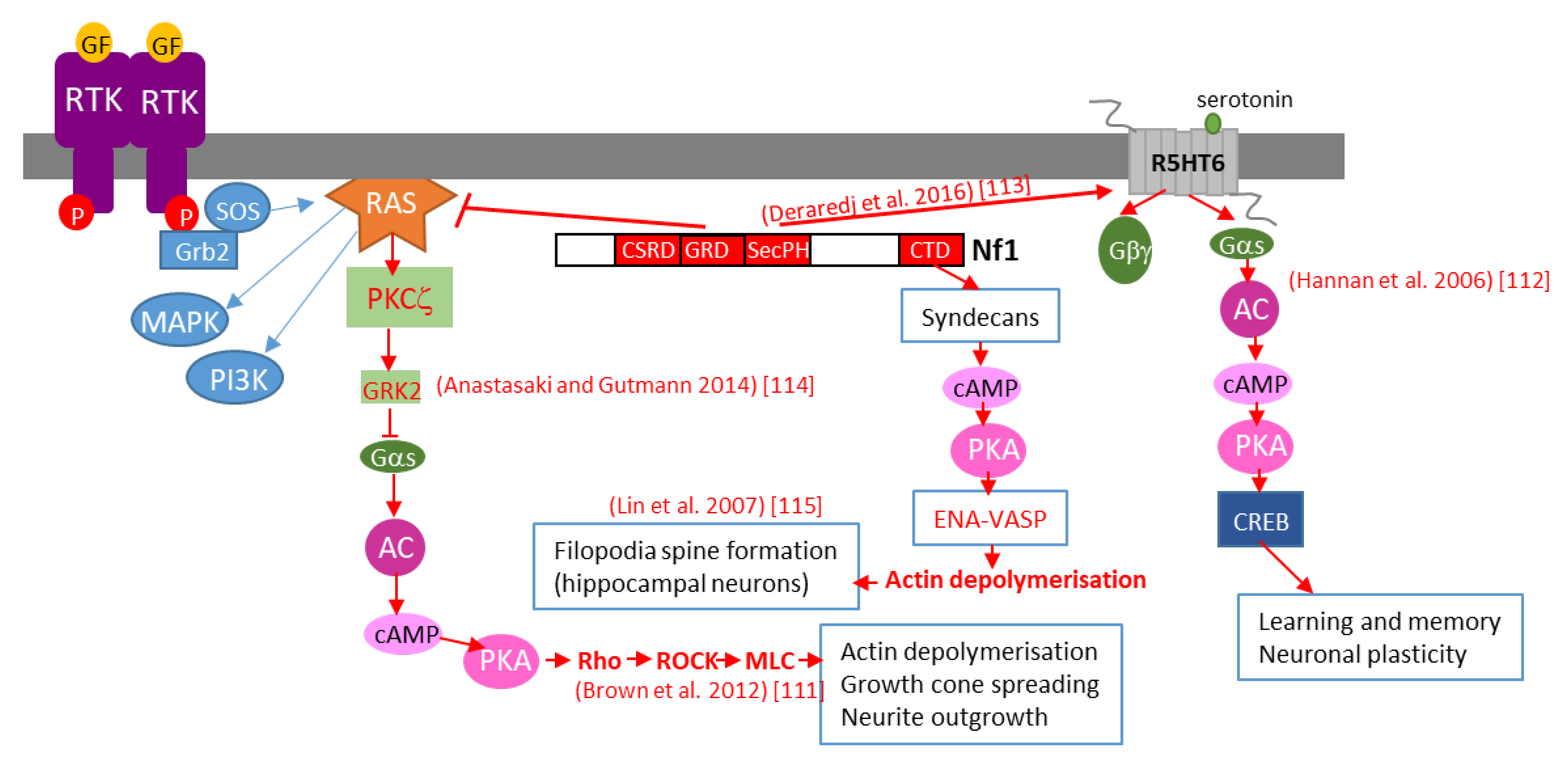
Figure 92. Schematic representation of various mechanisms of neurofibromin-mediated cAMP regulation. GF: growth factor, RTK: tyrosine kinase receptor, AC: adenylate cyclase, CREB: cAMP response element-binding protein, MAPK: mitogen-activated protein kinase, PI3K: phosphoinositide 3-kinase, SOS: son of sevenless, Grb2: growth factor receptor-bound protein 2.
Anastasaki and Gutmann (2014) [114][36] established a mechanism involving neurofibromin-dependent cAMP regulation in a Ras-dependent manner in hippocampal neurons. This process does not involve classical Ras-mediated MEK/AKT signaling but requires activation of an atypical PKC zeta by Ras, leading to GRK2-mediated Gαs inactivation [114][36] (Figure 92).
This pathway may represent the initial part of a more general pathway, described by Brown et al. (2012) [111][33], that was shown to regulate neurite growth and growth-cone formation in neurons of the CNS and involving PKA-dependent activation of the Rho/ROCK/MLC pathway (Figure 92).
We would not be exhaustive without citing the work of Lin et al. (2007) [115][37], who showed that neurofibromin is a binding partner of syndecan2, which induces syndecan2-dependant activation of PKA and its actin-associated downstream effectors Ena-VASP, thus inducing actin polymerization and dendritic filopodia formation and contributing to spinogenesis (Figure 92).
Unlike the examples given above, in Schwann cells, in which cAMP acts as a co-mitogen to many growth factors, neurofibromin negatively regulates cAMP. Mouse Schwann cells depleted of NF1 [101][23] and NF1 MPNST cell lines [116][38] show increased cAMP levels, which causes aberrant cell proliferation.
Overall, these data suggest that neurofibromin may act upstream of PKA by many ways to regulate cAMP production. On the other hand, it is known that PKA phosphorylates neurofibromin [54][39] and inhibits its GAP activity by allowing interaction of 14-3-3 with the neurofibromin CTD. It is thus possible that there is a negative feedback mechanism between neurofibromin and PKA. Furthermore, cAMP appears to be a potentially important therapeutic target to correct a set of phenotypes caused by loss or reduced expression of neurofibromin, especially learning defects in NF1-affected children.
3. Regulation of Dopamine Levels
Neurofibromin functions as a positive regulator of dopamine homeostasis. Indeed, NF1-mutant mice display reduced dopamine levels in the brain that lead to deficits in spatial learning, memory, and attention [117,118][40][41]. These manifestations were rescued by the administration of L-dopa or methylphenidate, thus supporting the use of methylphenidate to elevate dopamine levels in the treatment of children with NF1-associated learning and attention deficits [119][42]. Furthermore, a dose-dependent relationship between neurofibromin levels, dopamine signaling, and cognitive deficits was identified in the hippocampus and striatum of NF1 patients [120][43]. The molecular mechanisms responsible for such regulation are still unknown. Clinical trials conducted with methylphenidate showed an effect in the reduction of attention deficits, spatial working memory impairments, and ADHD symptoms in children with NF1 [121][44].
Recently, neurofibromin was shown to be involved in the dopamine-mediated production of cAMP in a population of striatal neurons (D2-dopamine receptor expressing medium spiny neurons), suggesting a role in motor learning [122][45].
4. Regulation of mTOR Signaling
Neurofibromin also regulates the mTOR pathway. This regulation takes place through the inactivation of the Ras/PI3K pathway, a growth factor-controlled upstream pathway that regulates mTOR (Figure 103).
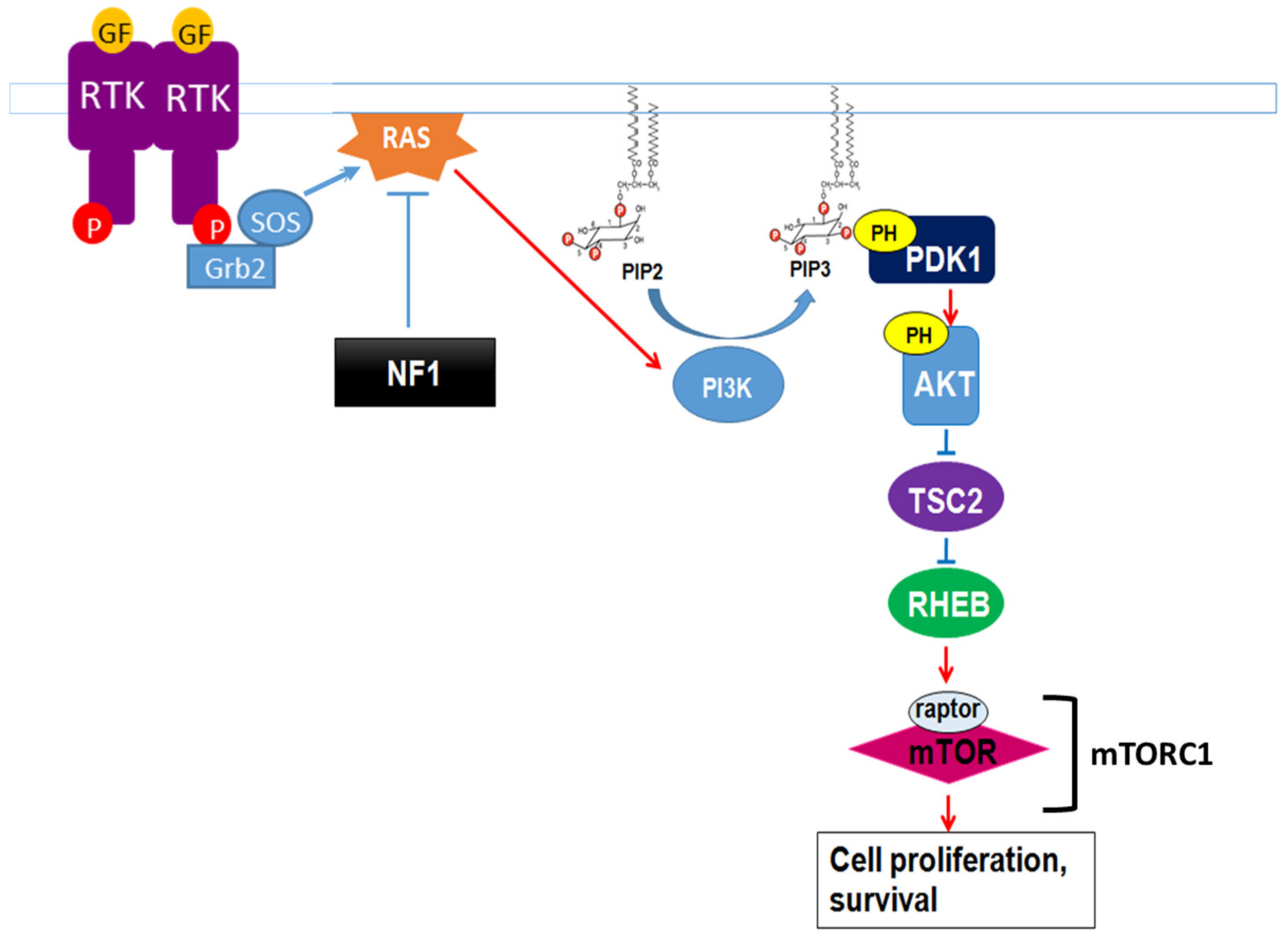
Figure 103. Neurofibromin regulation of the growth factor-controlled Ras/PI3K signaling pathway. PIP2: phosphatidylinositol-4,5-bisphosphate, PIP3: phosphatidylinositol 3,4,5 trisphosphate, PDK1: phosphoinositide-dependent kinase-1, PH: pleckstrin-homologous domain, TSC2: TSC complex subunit 2, RHEB: Ras homolog enriched in brain.
In the absence of neurofibromin, the hyperactivation of Ras leads to aberrant activation of mTOR via Akt-dependent phosphorylation and inactivation of TSC2, a GAP protein, negatively regulating the small GTPase Rheb. Indeed, mTOR is constitutively active in neurofibromin-deficient primary cells and tumor cell lines derived from NF1 patients. These cells are sensitive to the mTOR inhibitors rapamycin [123,124][46][47] and everolimus [125][48], suggesting that mTOR may constitute a therapeutic target for the treatment of NF1 tumors. Several clinical trials have been developed with different mTOR inhibitors against various manifestations of NF1, sometimes in combination with other drugs. Results were disappointing [126,127,128][49][50][51] except in the case of low-grade pediatric glioma, where a stabilization and a tumor shrinkage was observed [129][52].
In lysosomes, which constitute a hub for mTORC1 signaling, neurofibromin also regulates mTOR via another nutrient-controlled pathway. Indeed, in an effort to identify new neurofibromin partners using affinity purification followed by mass spectrometry (AP-MS), Li et al. (2017) [130][53] showed that neurofibromin directly interacts with LAMTOR1, a membrane protein localized to the surface of late endosomes and lysosomes that plays a role in the anchoring of the Ragulator complex (formed by five LAMTOR subunits) to these membranes. This Ragulator complex is crucial for mTORC1 activation on the lysosomal surface in response to amino acids [131][54]. Functional studies of this interaction demonstrated that neurofibromin negatively regulates mTORC1 signaling in a LAMTOR1-dependent manner [130][53] (Figure 114).
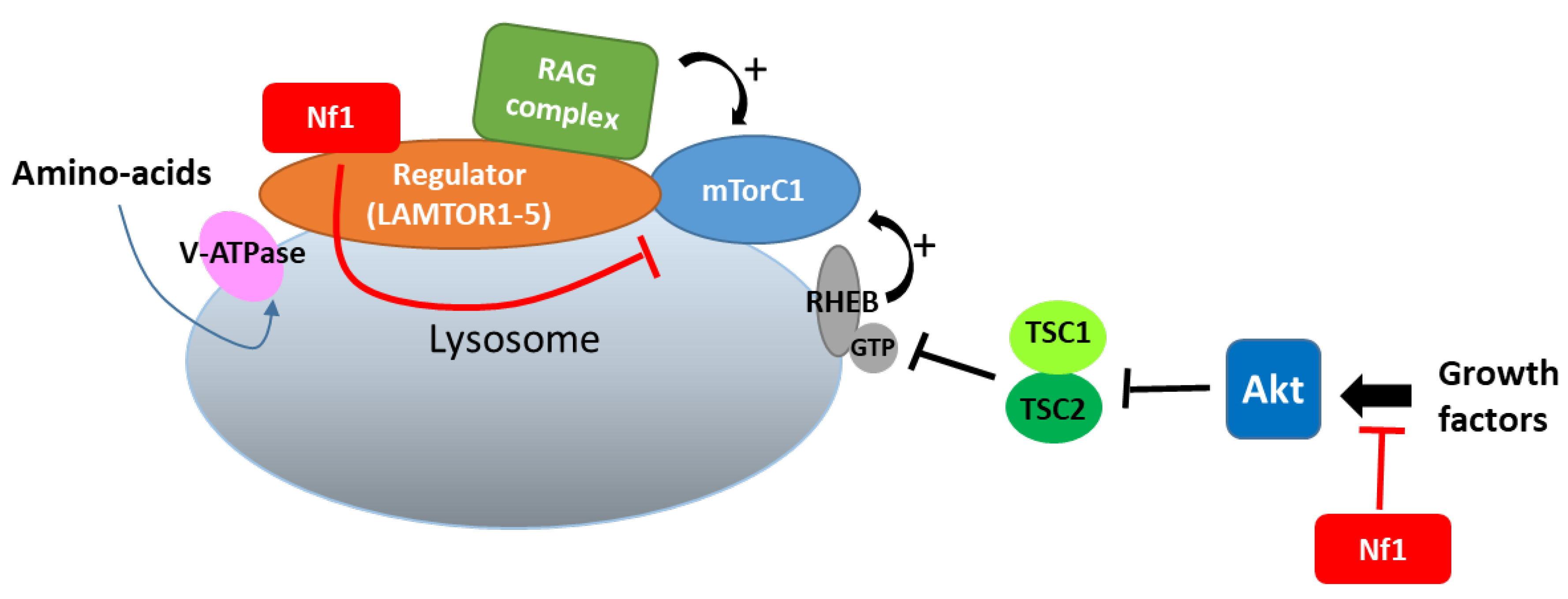
Figure 114.
Neurofibromin binds to LAMTOR1 and inhibits mTORC1 signaling.
Thus, neurofibromin negatively regulates the two complementary pathways (controlled by growth factors and amino acids), allowing the activation of mTORC1 in lysosomes.
Xie et al. (2016) [132][55] described a signaling mechanism in which GPCRs (opioid receptors) activate Ras-AKT-mTOR signaling in the striatum and showed that neurofibromin is required for the cross talk between GPCRs (opioid receptors) and Ras activation. Indeed, upon activation of opioid receptors by morphine, the released Gβγ subunits interact with the SecPH domain of neurofibromin and inhibit its Ras-GAP activity. This results in specific activation of Ras-AKT-mTOR signaling (Figure 125). Deletion of neurofibromin resulted in an increase in Ras baseline activity but abolished the opioid receptor-induced activation of Ras. This mechanism suggests a role for neurofibromin in drug addiction [132][55].
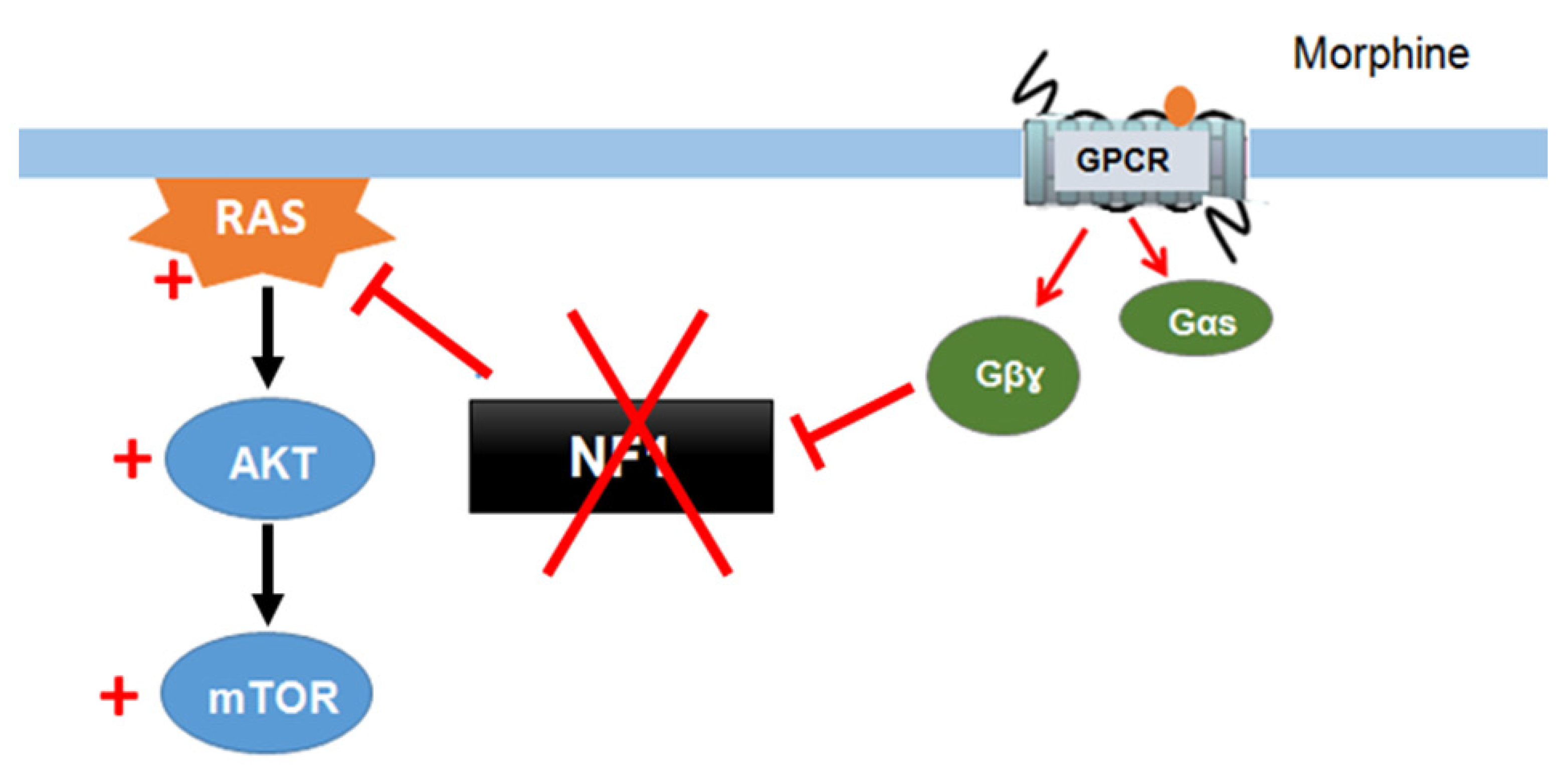
Figure 125. Neurofibromin mediates cross talk between GPCR (opioid receptors) and Ras/AKT signaling [132].
5. Control of Actin Cytoskeleton Organization
Actin exists in two forms, monomeric G-actin and polymeric F-actin. The fine-tuning of actin polymerization/depolymerization is important for the regulation of crucial biological processes, such as cell morphology and motility. Cofilin, a member of the ADF family (actin depolymerizing factor), depolymerizes actin by severing aged actin filaments. Phosphorylation of cofilin by LIM kinases (LIMK1 and LIMK2) inactivates its actin-severing activity, resulting in the stabilization of actin filaments, an increase in the number of stress fibers, and focal adhesion formation [133,134][56][57].
Aside from its Ras-GAP activity, neurofibromin plays an important role in regulating cytoskeletal organization. Specifically, neurofibromin regulates the dynamic reorganization and turnover of actin filaments through the negative regulation of two parallel pathways: the Rho/ROCK/LIMK2/cofilin and Rac1/Pak1/LIMK1/cofilin pathways. Using NF1 siRNA, Ozawa et al. (2005) [135][58] showed that depletion or down-regulation of NF1 activates the Rho/ROCK/LIMK2/cofilin signaling pathway and leads to the sustained phosphorylation and inactivation of cofilin by LIMK2, which in turn induces the formation of focal adhesions and stable actin stress fibers. The expression of neurofibromin-GRD type 2 (containing exon 23a), but not neurofibromin-GRD type 1, partially restores normal phosphorylated cofilin levels and suppresses the accumulation of actin stress fibers [135][58]. Consistent with a role of neurofibromin in cell adhesion and motility, Kweh et al. (2009) [61][59] demonstrated a physical interaction between the neurofibromin CTD and FAK and showed that NF1+/+ MEF cells adhere less to collagen I and fibronectin I than NF1−/− MEF cells and that the two cell genotypes displayed differences in the cellular distribution of actin and FAK [61][59].
Vallée et al. (2012) [136][60] described the molecular mechanism involved in the regulation of cytoskeletal dynamics through the Rho/ROCK/LIMK2/cofilin pathway by neurofibromin. Based on a yeast two-hybrid screening approach, they identified an interaction between neurofibromin SecPH and LIMK2. This interaction inhibits LIMK2 kinase activity on cofilin by preventing LIMK2 activation by ROCK, its upstream regulator (Figure 136).
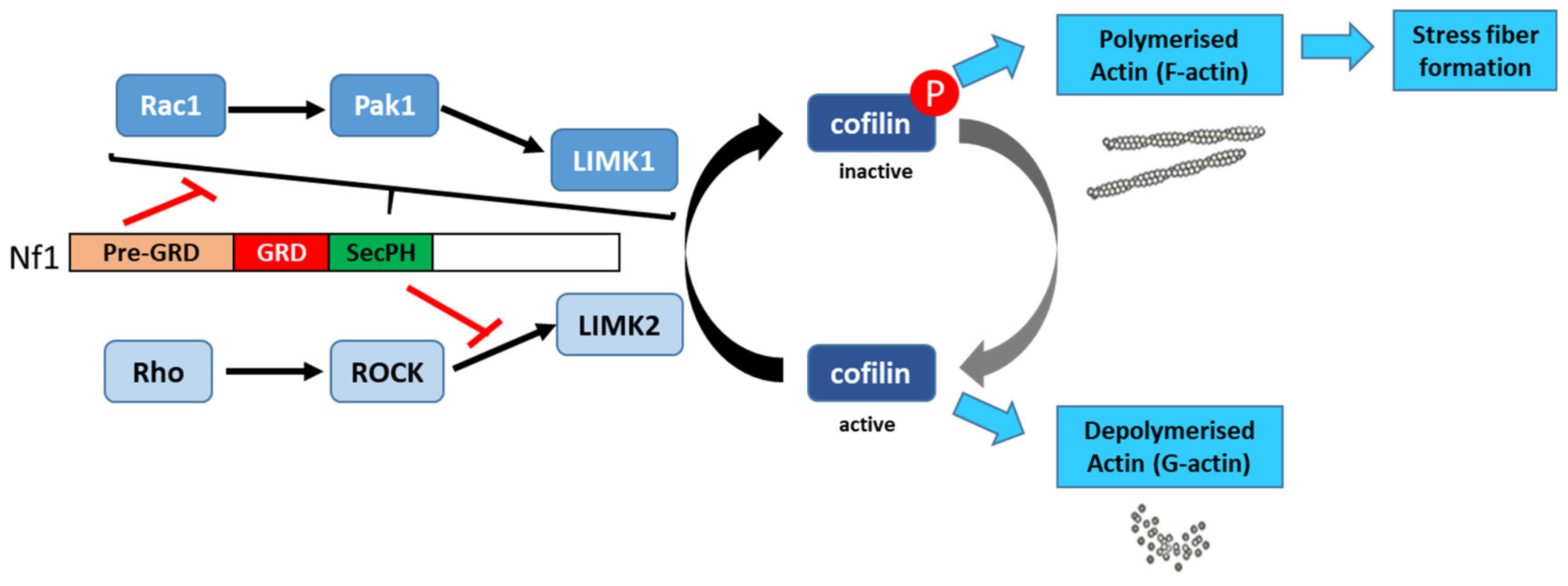
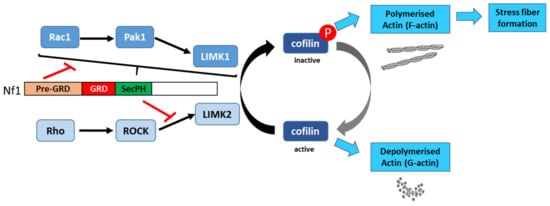
Thus, non-phosphorylated cofilin remains active and able to depolymerize actin, reducing LIMK2-induced actin stress-fiber accumulation [136][60]. On the other hand, neurofibromin inhibits the parallel signaling pathway centered on LIMK1. Indeed, the Rac1/Pak1/LIMK1/cofilin pathway is inhibited by the pre-GRD of neurofibromin (Nf11-1163) [137][61] (Figure 13). The pre-GRD was even hypothesized to possess direct Rac-GAP activity. The expression of Nf11-1163 in NF1-deficient cells significantly reduced stress-fiber formation and halted cell migration [137][61].
6. Microtubule-Dependent Transport in Melanocytes, Neurons, and Schwann Cells
Several independent studies have shown that neurofibromin directly interacts with or belongs to the same protein complex as various proteins involved in the microtubule-dependent transport of organelles, protein complexes, and mRNA in melanocytes, neurons, and Schwann cells. Indeed, in HeLa cells and calf brain, neurofibromin was shown to be in the same complex as kinesin-1, a motor protein involved in anterograde transport along microtubules [138][62]. De Sheppers et al. (2006) [81][63] further demonstrated a direct interaction between the neurofibromin GRD and amyloid precursor protein (APP) in melanosomes and APP was previously shown to directly interact with neuronal kinesin-1 and hypothesized to constitute a cargo receptor for kinesin-1 in neurons [139][64]. Similarly, Arun et al. (2013b) [140][65] further demonstrated an interaction between the neurofibromin TBD domain and the dynein heavy chain (DHC), a component of the dynein motor protein involved in retrograde transport along microtubules, in melanosomes. Overall, these data strongly suggest that neurofibromin plays an important role in the intracellular transport of melanosomes in melanocytes, which could account for the pathological mechanism of CALM (Café Au Lait Macule) formation. More generally, neurofibromin may play a role in neurotransmitter vesicle trafficking in neurons, which may be involved in cognitive disorders associated with NF1 disease.
Based on the comparison of gene expression in brain regions of WT and NF1+/− mice, Donarum et al. (2006) [141][66] further suggested a functional connection between neurofibromin, APP, and D3R (one of the five dopamine receptors) that interact with APP.
Other data obtained in Schwann cells showed that the neurofibromin TBD is able to interact with LRPPRC (leucine-rich pentatricopeptide repeat motif-containing protein) as part of a ribonucleoprotein complex connected to microtubules via kinesin-1 [142][67]. These complexes are consistent with RNA granules that contain mRNA and the ribosomal machinery to allow protein synthesis in response to appropriate cues in a temporal and spatial manner [143][68].
7. Cell Cycle
Independently of its Ras-GAP function, neurofibromin regulates the metaphase to anaphase transition. Neurofibromin is involved in the spindle assembly checkpoint (SAC) to induce mitotic arrest in response to spindle damage. This function is carried by the CTD, of which the overexpression in yeast delays transition between these two phases of the cell cycle [23][69].
Koliou et al. (2016) [58][70] showed that neurofibromin is localized to the mitotic spindle and that such localization may account for a role in regulating chromosome congression during mitosis. These authors further demonstrated that neurofibromin participates in spindle formation and proper chromosome metaphase alignment. Neurofibromin depletion led to aberrant chromosome congression at the metaphase plate, which typically caused chromosomal instability and aneuploidy [58][70].
8. Neurofibromin Interactions
The interacting partners of neurofibromin are summarized in Figure 147.
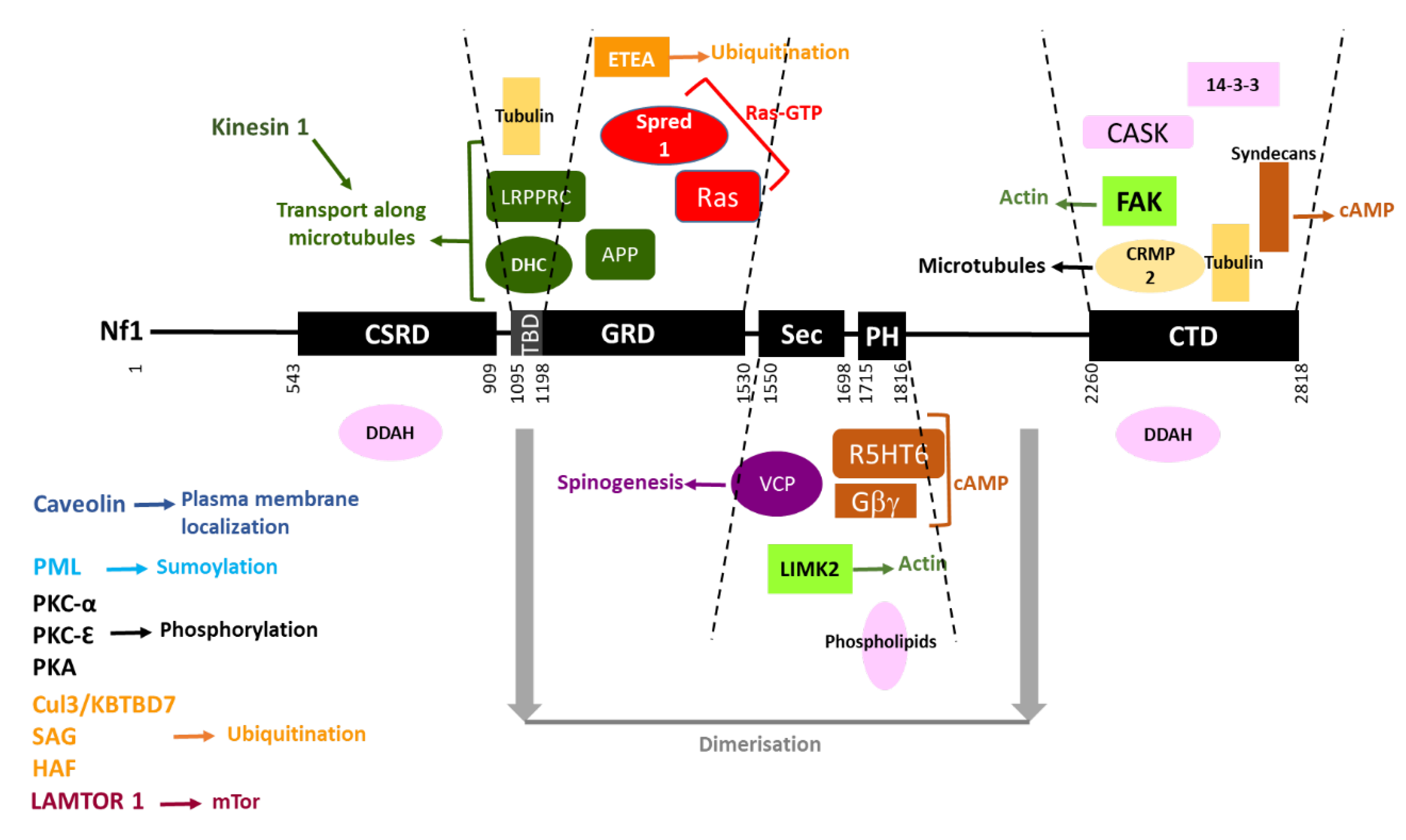
Figure 147. Neurofibromin interacting partners. Neurofibromin domains are indicated as black boxes and their limits are written below. Interacting partners of each domain are pictured between the dashed lines delimiting the domains. Each type of function is represented by a color: Ras-GTP in red, cAMP in brown, actin dynamics in green, microtubules in yellow, transport along microtubules in dark green, spinogenesis in purple, ubiquitination in orange, sumoylation in blue, phosphorylation in black, plasma membrane localization in dark blue, and unknown function in pink. Regions necessary for neurofibromin dimerization are indicated in grey. Partners interacting with unknown domains of neurofibromin or being part of the same complex are not pictured but their name is indicated on the left side of the figure.
References
- Xu, G.F.; Lin, B.; Tanaka, K.; Dunn, D.; Wood, D.; Gesteland, R.; White, R.; Weiss, R.; Tamanoi, F. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 1990, 63, 835–841.
- Martin, G.A.; Viskochil, D.; Bollag, G.; McCabe, P.C.; Crosier, W.J.; Haubruck, H.; Conroy, L.; Clark, R.; O’Connell, P.; Cawthon, R.M.; et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990, 63, 843–849.
- Ballester, R.; Marchuk, D.; Boguski, M.; Saulino, A.; Letcher, R.; Wigler, M.; Collins, F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990, 63, 851–859.
- Basu, T.N.; Gutmann, D.H.; Fletcher, J.A.; Glover, T.W.; Collins, F.S.; Downward, J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 1992, 356, 713–715.
- DeClue, J.E.; Papageorge, A.G.; Fletcher, J.A.; Diehl, S.R.; Ratner, N.; Vass, W.C.; Lowy, D.R. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 1992, 69, 265–273.
- Bollag, G.; McCormick, F. Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature 1991, 351, 576–579.
- Hiatt, K.K.; Ingram, D.A.; Zhang, Y.; Bollag, G.; Clapp, D.W. Neurofibromin GTPase-activating protein-related domains restore normal growth in Nf1-/- cells. J. Biol. Chem. 2001, 276, 7240–7245.
- Dasgupta, B.; Gutmann, D.H. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J. Neurosci. 2005, 25, 5584–5594.
- Oliveira, A.F.; Yasuda, R. Neurofibromin is the major ras inactivator in dendritic spines. J. Neurosci. 2014, 34, 776–783.
- Cichowski, K.; Santiago, S.; Jardim, M.; Johnson, B.W.; Jacks, T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes. Dev. 2003, 17, 449–454.
- Zhang, Y.Y.; Vik, T.A.; Ryder, J.W.; Srour, E.F.; Jacks, T.; Shannon, K.; Clapp, D.W. Nf1 regulates hematopoietic progenitor cell growth and ras signaling in response to multiple cytokines. J. Exp. Med. 1998, 187, 1893–1902.
- Hennig, A.; Markwart, R.; Wolff, K.; Schubert, K.; Cui, Y.; Prior, I.A.; Esparza-Franco, M.A.; Ladds, G.; Rubio, I. Feedback activation of neurofibromin terminates growth factor-induced Ras activation. Cell Commun. Signal. 2016, 14, 5.
- Dasgupta, B.; Li, W.; Perry, A.; Gutmann, D.H. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res. 2005, 65, 236–245.
- Widemann, B.C.; Dombi, E.; Gillespie, A.; Wolters, P.L.; Belasco, J.; Goldman, S.; Korf, B.R.; Solomon, J.; Martin, S.; Salzer, W.; et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro-Oncology 2014, 16, 707–718.
- Costa, R.M.; Federov, N.B.; Kogan, J.H.; Murphy, G.G.; Stern, J.; Ohno, M.; Kucherlapati, R.; Jacks, T.; Silva, A.J. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 2002, 415, 526–530.
- Li, W.; Cui, Y.; Kushner, S.A.; Brown, R.A.; Jentsch, J.D.; Frankland, P.W.; Cannon, T.D.; Silva, A.J. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr. Biol. 2005, 15, 1961–1967.
- Guilding, C.; McNair, K.; Stone, T.W.; Morris, B.J. Restored plasticity in a mouse model of neurofibromatosis type 1 via inhibition of hyperactive ERK and CREB. Eur. J. Neurosci. 2007, 25, 99–105.
- Molosh, A.I.; Johnson, P.L.; Spence, J.P.; Arendt, D.; Federici, L.M.; Bernabe, C.; Janasik, S.P.; Segu, Z.M.; Khanna, R.; Goswami, C.; et al. Social learning and amygdala disruptions in Nf1 mice are rescued by blocking p21-activated kinase. Nat. Neurosci. 2014, 17, 1583–1590.
- Krab, L.C.; de Goede-Bolder, A.; Aarsen, F.K.; Pluijm, S.M.; Bouman, M.J.; van der Geest, J.N.; Lequin, M.; Catsman, C.E.; Arts, W.F.; Kushner, S.A.; et al. Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: A randomized controlled trial. JAMA 2008, 300, 287–294.
- Van der Vaart, T.; Plasschaert, E.; Rietman, A.B.; Renard, M.; Oostenbrink, R.; Vogels, A.; de Wit, M.C.; Descheemaeker, M.J.; Vergouwe, Y.; Catsman-Berrevoets, C.E.; et al. Simvastatin for cognitive deficits and behavioural problems in patients with neurofibromatosis type 1 (NF1-SIMCODA): A randomised, placebo-controlled trial. Lancet Neurol. 2013, 12, 1076–1083.
- Payne, J.M.; Barton, B.; Ullrich, N.J.; Cantor, A.; Hearps, S.J.; Cutter, G.; Rosser, T.; Walsh, K.S.; Gioia, G.A.; Wolters, P.L.; et al. Randomized placebo-controlled study of lovastatin in children with neurofibromatosis type 1. Neurology 2016, 87, 2575–2584.
- Cui, Y.; Costa, R.M.; Murphy, G.G.; Elgersma, Y.; Zhu, Y.; Gutmann, D.H.; Parada, L.F.; Mody, I.; Silva, A.J. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 2008, 135, 549–560.
- Kim, H.A.; Ratner, N.; Roberts, T.M.; Stiles, C.D. Schwann cell proliferative responses to cAMP and Nf1 are mediated by cyclin D1. J. Neurosci. 2001, 21, 1110–1116.
- Warrington, N.M.; Gianino, S.M.; Jackson, E.; Goldhoff, P.; Garbow, J.R.; Piwnica-Worms, D.; Gutmann, D.H.; Rubin, J.B. Cyclic AMP suppression is sufficient to induce gliomagenesis in a mouse model of neurofibromatosis-1. Cancer Res. 2010, 70, 5717–5727.
- Guo, H.F.; The, I.; Hannan, F.; Bernards, A.; Zhong, Y. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science 1997, 276, 795–798.
- Wolman, M.A.; de Groh, E.D.; McBride, S.M.; Jongens, T.A.; Granato, M.; Epstein, J.A. Modulation of cAMP and ras signaling pathways improves distinct behavioral deficits in a zebrafish model of neurofibromatosis type 1. Cell Rep. 2014, 8, 1265–1270.
- Guo, H.F.; Tong, J.; Hannan, F.; Luo, L.; Zhong, Y. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature 2000, 403, 895–898.
- The, I.; Hannigan, G.E.; Cowley, G.S.; Reginald, S.; Zhong, Y.; Gusella, J.F.; Hariharan, I.K.; Bernards, A. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science 1997, 276, 791–794.
- Tong, J.; Hannan, F.; Zhu, Y.; Bernards, A.; Zhong, Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat. Neurosci. 2002, 5, 95–96.
- Dasgupta, B.; Gutmann, D.H. Neurofibromatosis 1: Closing the GAP between mice and men. Curr. Opin. Genet. Dev. 2003, 13, 20–27.
- Hegedus, B.; Dasgupta, B.; Shin, J.E.; Emnett, R.J.; Hart-Mahon, E.K.; Elghazi, L.; Bernal-Mizrachi, E.; Gutmann, D.H. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell 2007, 1, 443–457.
- Brown, J.A.; Gianino, S.M.; Gutmann, D.H. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J. Neurosci. 2010, 30, 5579–5589.
- Brown, J.A.; Diggs-Andrews, K.A.; Gianino, S.M.; Gutmann, D.H. Neurofibromatosis-1 heterozygosity impairs CNS neuronal morphology in a cAMP/PKA/ROCK-dependent manner. Mol. Cell Neurosci. 2012, 49, 13–22.
- Hannan, F.; Ho, I.; Tong, J.J.; Zhu, Y.; Nurnberg, P.; Zhong, Y. Effect of neurofibromatosis type I mutations on a novel pathway for adenylyl cyclase activation requiring neurofibromin and Ras. Hum. Mol. Genet. 2006, 15, 1087–1098.
- Deraredj Nadim, W.; Chaumont-Dubel, S.; Madouri, F.; Cobret, L.; De Tauzia, M.L.; Zajdel, P.; Bénédetti, H.; Marin, P.; Morisset-Lopez, S. Physical interaction between neurofibromin and serotonin 5-HT6 receptor promotes receptor constitutive activity. Proc. Natl. Acad. Sci. USA 2016, 113, 12310–12315.
- Anastasaki, C.; Gutmann, D.H. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum. Mol. Genet. 2014, 23, 6712–6721.
- Lin, Y.L.; Lei, Y.T.; Hong, C.J.; Hsueh, Y.P. Syndecan-2 induces filopodia and dendritic spine formation via the neurofibromin-PKA-Ena/VASP pathway. J. Cell Biol. 2007, 177, 829–841.
- Dang, I.; De Vries, G.H. Aberrant cAMP metabolism in NF1 malignant peripheral nerve sheath tumor cells. Neurochem. Res. 2011, 36, 1697–1705.
- Izawa, I.; Tamaki, N.; Saya, H. Phosphorylation of neurofibromatosis type 1 gene product (neurofibromin) by cAMP-dependent protein kinase. FEBS Lett. 1996, 382, 53–59.
- Brown, J.A.; Emnett, R.J.; White, C.R.; Yuede, C.M.; Conyers, S.B.; O’Malley, K.L.; Wozniak, D.F.; Gutmann, D.H. Reduced striatal dopamine underlies the attention system dysfunction in neurofibromatosis-1 mutant mice. Hum. Mol. Genet. 2010, 19, 4515–4528.
- Diggs-Andrews, K.A.; Tokuda, K.; Izumi, Y.; Zorumski, C.F.; Wozniak, D.F.; Gutmann, D.H. Dopamine deficiency underlies learning deficits in neurofibromatosis-1 mice. Ann. Neurol. 2013, 73, 309–315.
- Mautner, V.F.; Kluwe, L.; Thakker, S.D.; Leark, R.A. Treatment of ADHD in neurofibromatosis type 1. Dev. Med. Child Neurol. 2002, 44, 164–170.
- Anastasaki, C.; Woo, A.S.; Messiaen, L.M.; Gutmann, D.H. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum. Mol. Genet. 2015, 24, 3518–3528.
- Pride, N.A.; Barton, B.; Hutchins, P.; Coghill, D.R.; Korgaonkar, M.S.; Hearps, S.J.C.; Rouel, M.; Malarbi, S.; North, K.N.; Payne, J.M. Effects of methylphenidate on cognition and behaviour in children with neurofibromatosis type 1: A study protocol for a randomised placebo-controlled crossover trial. BMJ Open 2018, 8, e021800.
- Sutton, L.P.; Muntean, B.S.; Ostrovskaya, O.; Zucca, S.; Dao, M.; Orlandi, C.; Song, C.; Xie, K.; Martemyanov, K.A. NF1-cAMP signaling dissociates cell type-specific contributions of striatal medium spiny neurons to reward valuation and motor control. PLoS Biol. 2019, 17, e3000477.
- Johannessen, C.M.; Reczek, E.E.; James, M.F.; Brems, H.; Legius, E.; Cichowski, K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc. Natl. Acad. Sci. USA 2005, 102, 8573–8578.
- Dasgupta, B.; Yi, Y.; Chen, D.Y.; Weber, J.D.; Gutmann, D.H. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005, 65, 2755–2760.
- Endo, M.; Yamamoto, H.; Setsu, N.; Kohashi, K.; Takahashi, Y.; Ishii, T.; Iida, K.; Matsumoto, Y.; Hakozaki, M.; Aoki, M.; et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2013, 19, 450–461.
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.A.; Cantor, A.; Korf, B.; Perentesis, J.; Gutmann, D.H.; Schorry, E.; et al. Sirolimus for non-progressive NF1-associated plexiform neurofibromas: An NF clinical trials consortium phase II study. Pediatr. Blood Cancer 2014, 61, 982–986.
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.; Cantor, A.; Perentesis, J.; Schorry, E.; Ullrich, N.; Gutmann, D.H.; et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: A neurofibromatosis Clinical Trials Consortium phase II study. Neuro-Oncology 2015, 17, 596–603.
- Widemann, B.C.; Lu, Y.; Reinke, D.; Okuno, S.H.; Meyer, C.F.; Cote, G.M.; Chugh, R.; Milhem, M.M.; Hirbe, A.C.; Kim, A.; et al. Targeting Sporadic and Neurofibromatosis Type 1 (NF1) Related Refractory Malignant Peripheral Nerve Sheath Tumors (MPNST) in a Phase II Study of Everolimus in Combination with Bevacizumab (SARC016). Sarcoma 2019, 2019, 7656747.
- Ullrich, N.J.; Prabhu, S.P.; Reddy, A.T.; Fisher, M.J.; Packer, R.; Goldman, S.; Robison, N.J.; Gutmann, D.H.; Viskochil, D.H.; Allen, J.C.; et al. A phase II study of continuous oral mTOR inhibitor everolimus for recurrent, radiographic-progressive neurofibromatosis type 1-associated pediatric low-grade glioma: A Neurofibromatosis Clinical Trials Consortium study. Neuro-Oncology 2020, 22, 1527–1535.
- Li, X.; Gao, M.; Choi, J.M.; Kim, B.J.; Zhou, M.T.; Chen, Z.; Jain, A.N.; Jung, S.Y.; Yuan, J.; Wang, W.; et al. Clustered, Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-coupled Affinity Purification/Mass Spectrometry Analysis Revealed a Novel Role of Neurofibromin in mTOR Signaling. Mol. Cell Proteom. 2017, 16, 594–607.
- Nada, S.; Mori, S.; Takahashi, Y.; Okada, M. p18/LAMTOR1: A late endosome/lysosome-specific anchor protein for the mTORC1/MAPK signaling pathway. Methods Enzymol. 2014, 535, 249–263.
- Xie, K.; Colgan, L.A.; Dao, M.T.; Muntean, B.S.; Sutton, L.P.; Orlandi, C.; Boye, S.L.; Boye, S.E.; Shih, C.C.; Li, Y.; et al. NF1 Is a Direct G Protein Effector Essential for Opioid Signaling to Ras in the Striatum. Curr. Biol. 2016, 26, 2992–3003.
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465.
- Scott, R.W.; Olson, M.F. LIM kinases: Function, regulation and association with human disease. J. Mol. Med. 2007, 85, 555–568.
- Ozawa, T.; Araki, N.; Yunoue, S.; Tokuo, H.; Feng, L.; Patrakitkomjorn, S.; Hara, T.; Ichikawa, Y.; Matsumoto, K.; Fujii, K.; et al. The neurofibromatosis type 1 gene product neurofibromin enhances cell motility by regulating actin filament dynamics via the Rho-ROCK-LIMK2-cofilin pathway. J. Biol. Chem. 2005, 280, 39524–39533.
- Kweh, F.; Zheng, M.; Kurenova, E.; Wallace, M.; Golubovskaya, V.; Cance, W.G. Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol. Carcinog. 2009, 48, 1005–1017.
- Vallée, B.; Doudeau, M.; Godin, F.; Gombault, A.; Tchalikian, A.; de Tauzia, M.L.; Bénédetti, H. Nf1 RasGAP inhibition of LIMK2 mediates a new cross-talk between Ras and Rho pathways. PLoS ONE 2012, 7, e47283.
- Starinsky-Elbaz, S.; Faigenbloom, L.; Friedman, E.; Stein, R.; Kloog, Y. The pre-GAP-related domain of neurofibromin regulates cell migration through the LIM kinase/cofilin pathway. Mol. Cell Neurosci. 2009, 42, 278–287.
- Hakimi, M.A.; Speicher, D.W.; Shiekhattar, R. The motor protein kinesin-1 links neurofibromin and merlin in a common cellular pathway of neurofibromatosis. J. Biol. Chem. 2002, 277, 36909–36912.
- De Schepper, S.; Boucneau, J.M.; Westbroek, W.; Mommaas, M.; Onderwater, J.; Messiaen, L.; Naeyaert, J.M.; Lambert, J.L. Neurofibromatosis type 1 protein and amyloid precursor protein interact in normal human melanocytes and colocalize with melanosomes. J. Investgi. Dermatol. 2006, 126, 653–659.
- Kamal, A.; Stokin, G.B.; Yang, Z.; Xia, C.H.; Goldstein, L.S. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 2000, 28, 449–459.
- Arun, V.; Worrell, L.; Wiley, J.C.; Kaplan, D.R.; Guha, A. Neurofibromin interacts with the cytoplasmic Dynein Heavy Chain 1 in melanosomes of human melanocytes. FEBS Lett. 2013, 587, 1466–1473.
- Donarum, E.A.; Halperin, R.F.; Stephan, D.A.; Narayanan, V. Cognitive dysfunction in Nf1 knock-out mice may result from altered vesicular trafficking of APP/DRD3 complex. BMC Neurosci. 2006, 7, 22.
- Arun, V.; Wiley, J.C.; Kaur, H.; Kaplan, D.R.; Guha, A. A novel neurofibromin (NF1) interaction with the leucine-rich pentatricopeptide repeat motif-containing protein links neurofibromatosis type 1 and the French Canadian variant of Leigh’s syndrome in a common molecular complex. J. Neurosci. Res. 2013, 91, 494–505.
- Wang, W.; van Niekerk, E.; Willis, D.E.; Twiss, J.L. RNA transport and localized protein synthesis in neurological disorders and neural repair. Dev. Neurobiol. 2007, 67, 1166–1182.
- Luo, G.; Kim, J.; Song, K. The C-terminal domains of human neurofibromin and its budding yeast homologs Ira1 and Ira2 regulate the metaphase to anaphase transition. Cell Cycle 2014, 13, 2780–2789.
- Koliou, X.; Fedonidis, C.; Kalpachidou, T.; Mangoura, D. Nuclear import mechanism of neurofibromin for localization on the spindle and function in chromosome congression. J. Neurochem. 2016, 136, 78–91.
More