Bacteriophages (phages), as natural antibacterial agents, are being rediscovered because of the growing threat of multi- and pan-drug-resistant bacterial pathogens globally. Most phages package their genome in the proteinaceous capsid (or head) and have a tail attached to the capsid. Tailed double-stranded DNA bacteriophages belonging to the class Caudoviricetes (Cauda means “tail” in Latin) are the most prevalent (~96% of all known phages). Based on tail morphology, they are further classified into three morphotypes: myovirus, siphovirus, and podovirus. Myophages (e.g., T4, T2, Mu, S16, and φKZ) have long, rigid, contractile tails with a sheath around a central tube; siphophages (e.g., λ, T5, HK97, and SPP1) possess long, flexible, non-contractile tails; and podophages (e.g., T7, T3, P22, and φ29) have short, non-contractile tails. Of these, myophages possess the most complex tail architectures with the greatest number of proteins involved in tail assembly and function.
- bacteriophage (phage)
- T4 phage
- tail fiber
- tail fiber structure
1. Molecular and Structural Insight of the Interaction between T4 Phage Long Tail Fibers (LTF) and Escherichia coli Receptors
1.1. T4 Phage Architecture and the Structure of Long Tail Fibers (LTF)
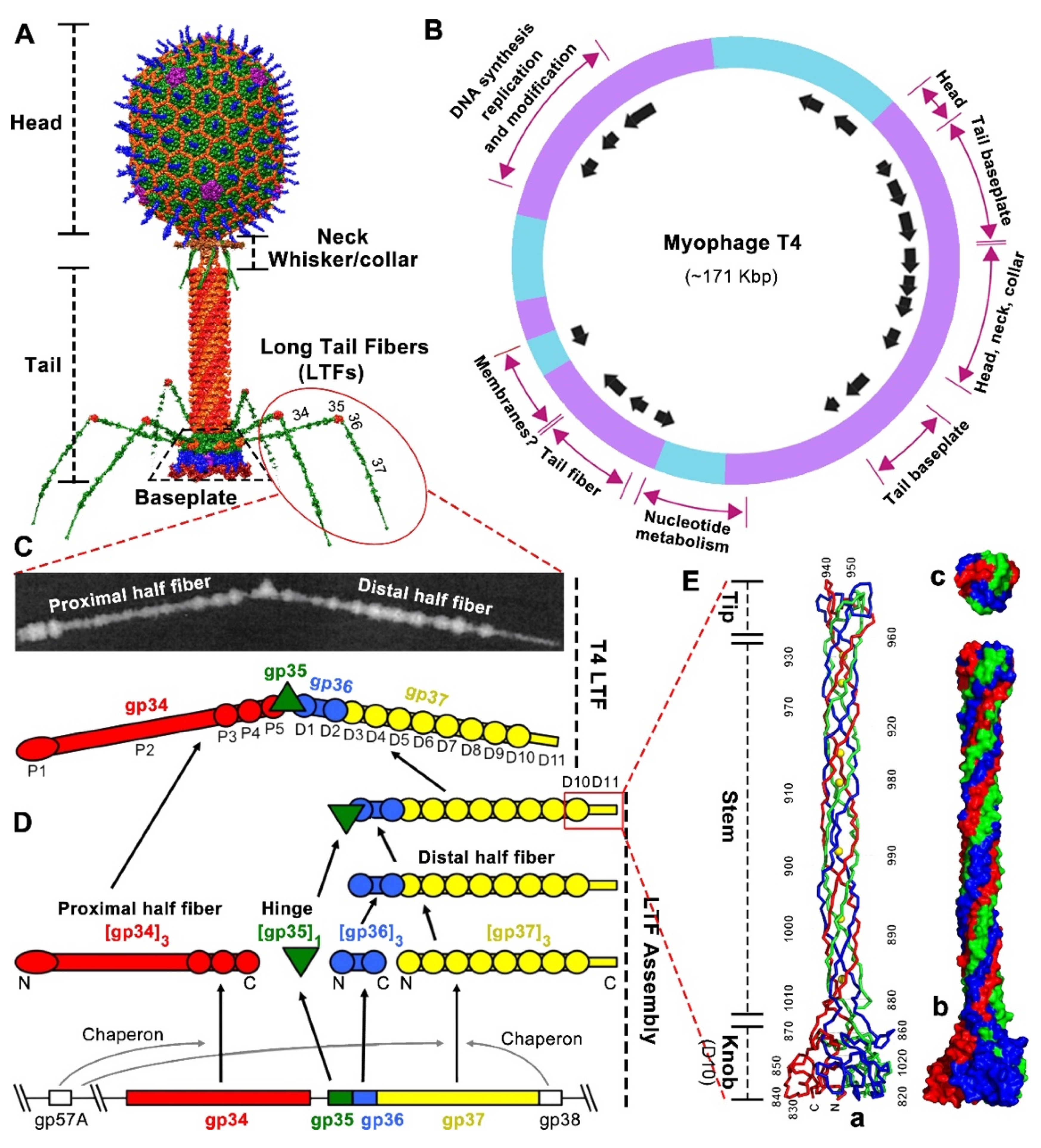
1.2. Molecular and Structural Insight of T4 LTFs’ Interaction with Host Receptors LPS and OmpC
1.2.1. T4 LTF “Tip” Binding to LPS
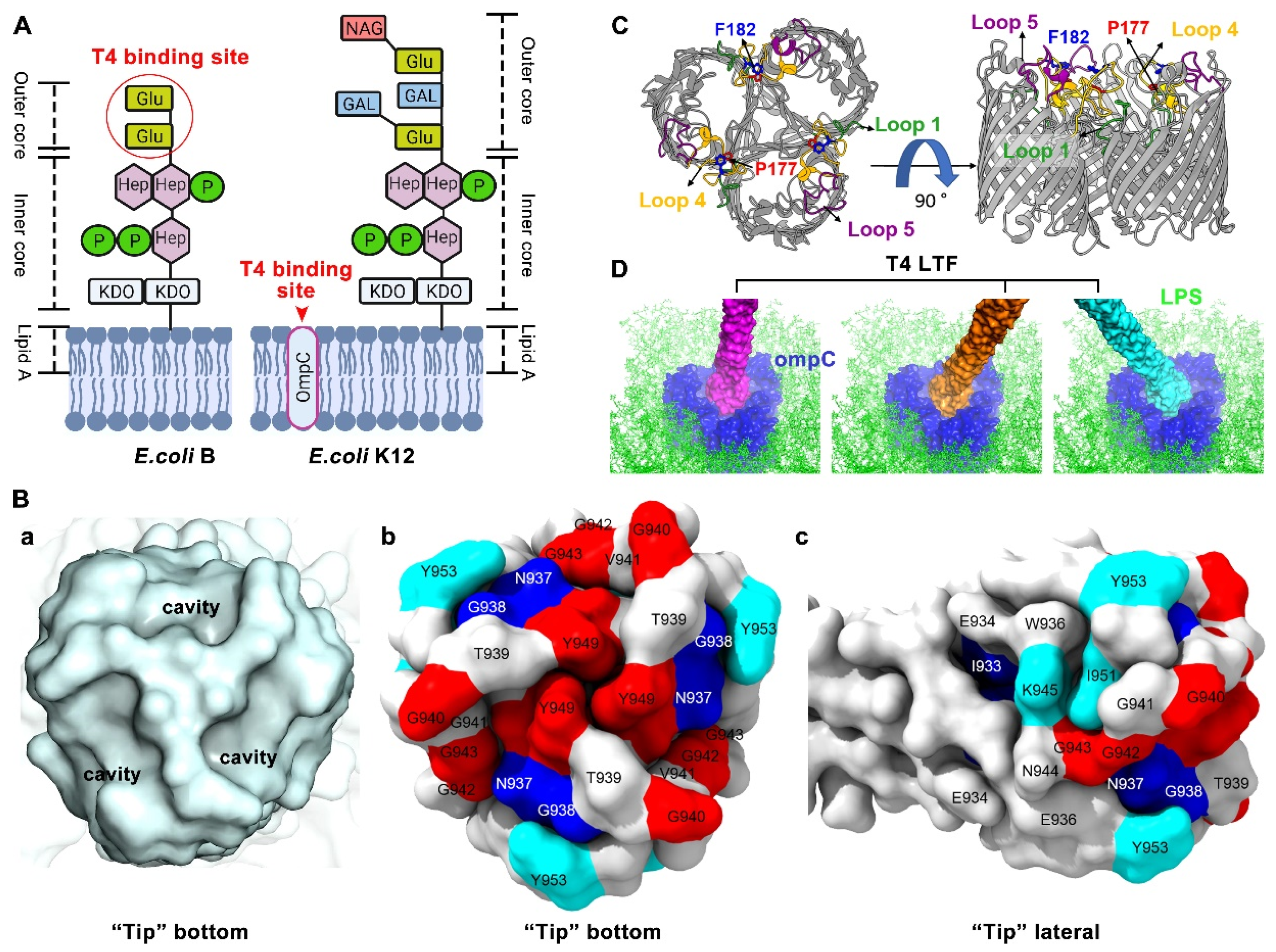
1.2.2. T4 LTF “Tip” Binding to OmpC
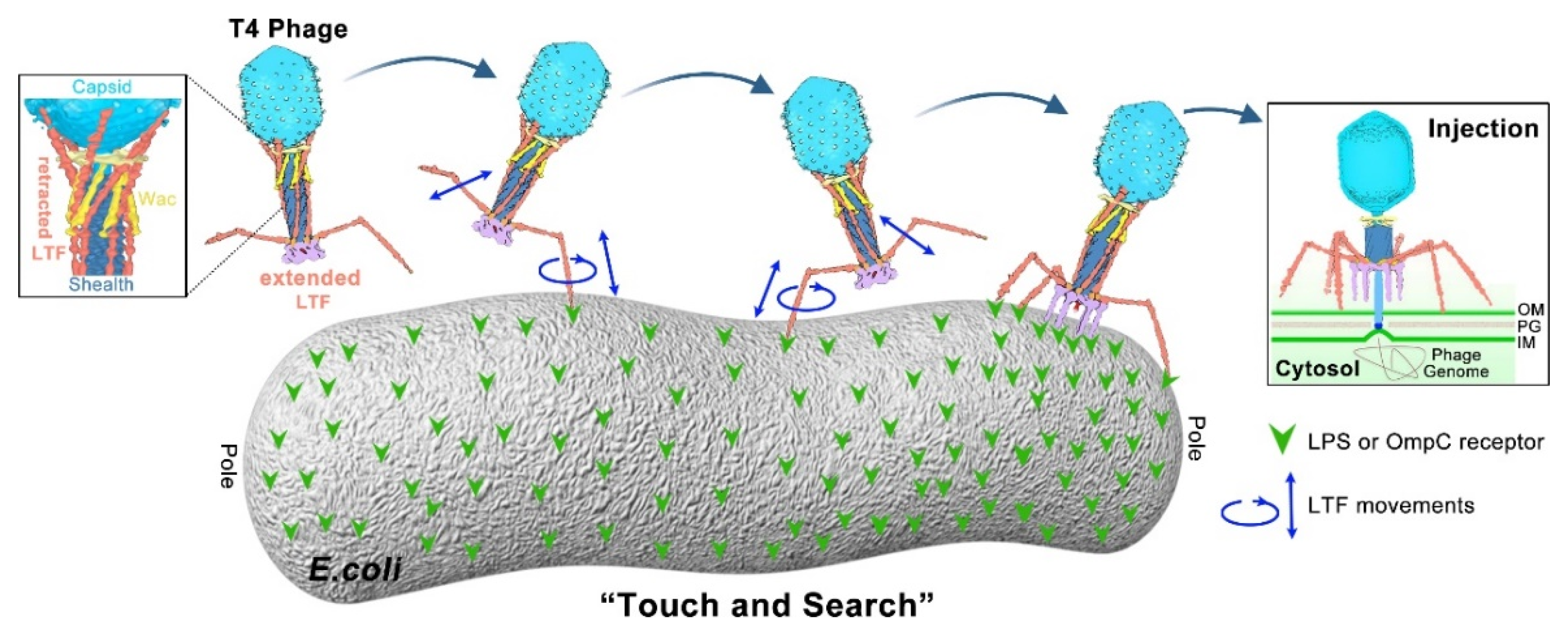
1.3. Model of Long Tail Fibers (LTFs) during T4 Infection Initiation
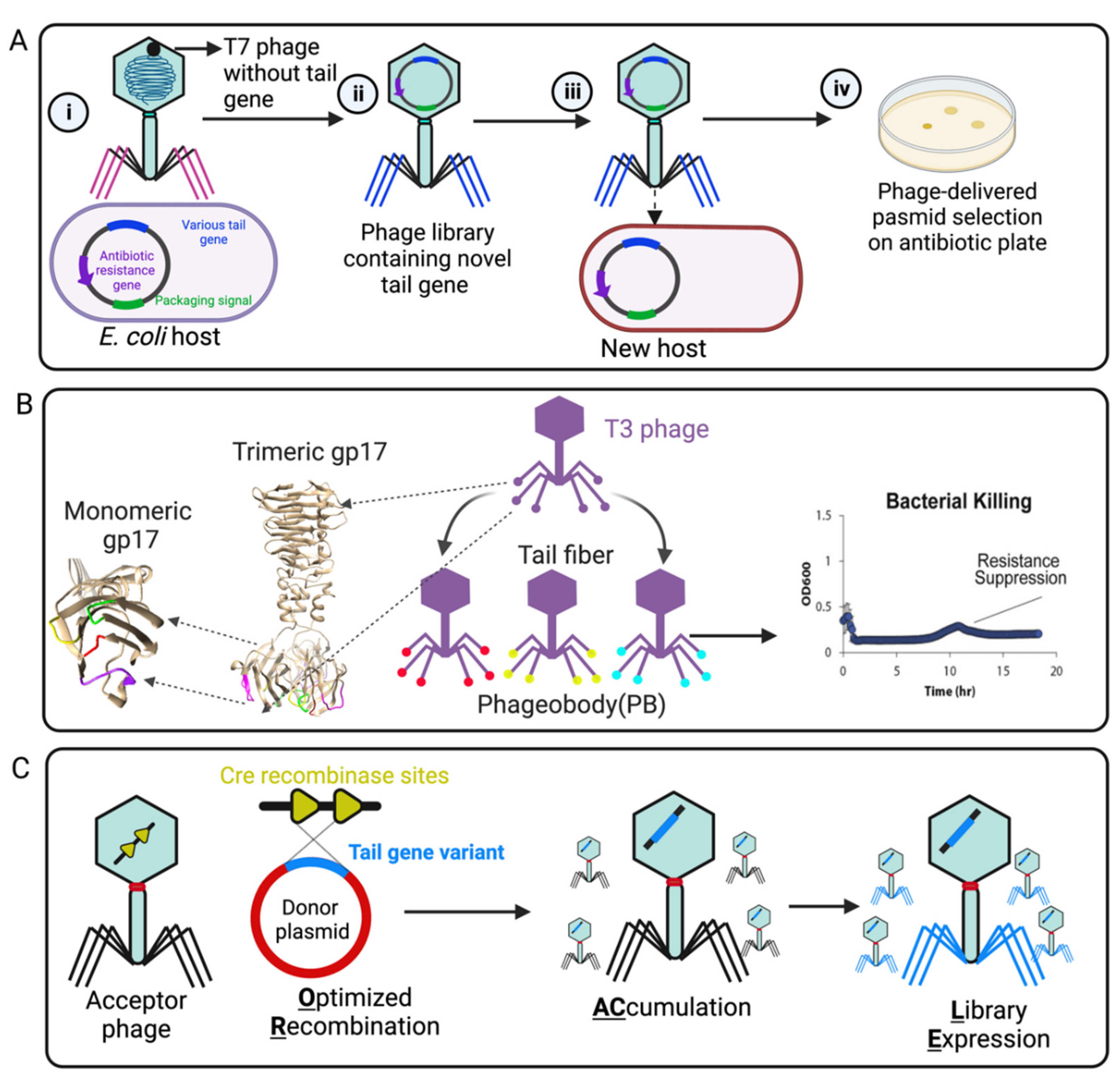
2. Engineering Strategies of Phage Tail Fiber for Reprogramming Phage Host Range
2.1. Host Range Widening through Natural Evolution
2.2. Rational Genetic Engineering of Tail Fibers
2.3. Bioinformatic Prediction of Phage Host Range
References
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; Curtis Hendrickson, R.; et al. Recent changes to virus taxonomy ratified by the International Committee on Taxonomy of Viruses (2022). Arch. Virol. 2022.
- Tétart, F.; Repoila, F.; Monod, C.; Krisch, H.M. Bacteriophage T4 Host Range is Expanded by Duplications of a Small Domain of the Tail Fiber Adhesin. J. Mol. Biol. 1996, 258, 726–731.
- Rao, V.B.; Black, L.W. Structure and assembly of bacteriophage T4 head. Virol. J. 2010, 7, 356.
- Chen, Z.; Sun, L.; Zhang, Z.; Fokine, A.; Padilla-Sanchez, V.; Hanein, D.; Jiang, W.; Rossmann, M.G.; Rao, V.B. Cryo-EM structure of the bacteriophage T4 isometric head at 3.3-Å resolution and its relevance to the assembly of icosahedral viruses. Proc. Natl. Acad. Sci. USA 2017, 114, E8184–E8193.
- Fokine, A.; Chipman, P.R.; Leiman, P.G.; Mesyanzhinov, V.V.; Rao, V.B.; Rossmann, M.G. Molecular architecture of the prolate head of bacteriophage T4. Proc. Natl. Acad. Sci. USA 2004, 101, 6003–6008.
- Zhu, J.; Tao, P.; Mahalingam, M.; Sha, J.; Kilgore, P.; Chopra, A.K.; Rao, V. A prokaryotic-eukaryotic hybrid viral vector for delivery of large cargos of genes and proteins into human cells. Sci. Adv. 2019, 5, eaax0064.
- Zhu, J.; Ananthaswamy, N.; Jain, S.; Batra, H.; Tang, W.-C.; Lewry, D.A.; Richards, M.L.; David, S.A.; Kilgore, P.B.; Sha, J.; et al. A universal bacteriophage T4 nanoparticle platform to design multiplex SARS-CoV-2 vaccine candidates by CRISPR engineering. Sci. Adv. 2021, 7, eabh1547.
- Zhu, J.; Jain, S.; Sha, J.; Batra, H.; Ananthaswamy, N.; Paul, B.K.; Emily, K.H.; Yashoda, M.H.; Wu, X.; Juan, P.O.; et al. A Bacteriophage-Based, Highly Efficacious, Needle- and Adjuvant-Free, Mucosal COVID-19 Vaccine. mBio 2022, 13, e01822-22.
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Ruger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156.
- Wu, X.; Zhu, J.; Tao, P.; Rao, V.B. Bacteriophage T4 Escapes CRISPR Attack by Minihomology Recombination and Repair. mBio 2021, 12, e0136121.
- Comeau, A.M.; Bertrand, C.; Letarov, A.; Tétart, F.; Krisch, H.M. Modular architecture of the T4 phage superfamily: A conserved core genome and a plastic periphery. Virology 2007, 362, 384–396.
- Sun, L.; Zhang, X.; Gao, S.; Rao, P.A.; Padilla-Sanchez, V.; Chen, Z.; Sun, S.; Xiang, Y.; Subramaniam, S.; Rao, V.B.; et al. Cryo-EM structure of the bacteriophage T4 portal protein assembly at near-atomic resolution. Nat. Commun. 2015, 6, 7548.
- Fokine, A.; Zhang, Z.; Kanamaru, S.; Bowman, V.D.; Aksyuk, A.A.; Arisaka, F.; Rao, V.B.; Rossmann, M.G. The molecular architecture of the bacteriophage T4 neck. J. Mol. Biol. 2013, 425, 1731–1744.
- Kostyuchenko, V.A.; Chipman, P.R.; Leiman, P.G.; Arisaka, F.; Mesyanzhinov, V.V.; Rossmann, M.G. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat. Struct. Mol. Biol. 2005, 12, 810–813.
- Letarov, A.; Manival, X.; Desplats, C.; Krisch, H.M. gpwac of the T4-type bacteriophages: Structure, function, and evolution of a segmented coiled-coil protein that controls viral infectivity. J. Bacteriol. 2005, 187, 1055–1066.
- Taylor, N.M.; Prokhorov, N.S.; Guerrero-Ferreira, R.C.; Shneider, M.M.; Browning, C.; Goldie, K.N.; Stahlberg, H.; Leiman, P.G. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 2016, 533, 346–352.
- Yap, M.L.; Klose, T.; Arisaka, F.; Speir, J.A.; Veesler, D.; Fokine, A.; Rossmann, M.G. Role of bacteriophage T4 baseplate in regulating assembly and infection. Proc. Natl. Acad. Sci. USA 2016, 113, 2654–2659.
- Arisaka, F.; Yap, M.L.; Kanamaru, S.; Rossmann, M.G. Molecular assembly and structure of the bacteriophage T4 tail. Biophys. Rev. 2016, 8, 385–396.
- Hyman, P.; van Raaij, M. Bacteriophage T4 long tail fiber domains. Biophys. Rev. 2017, 10, 463–471.
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. Structural remodeling of bacteriophage T4 and host membranes during infection initiation. Proc. Natl. Acad. Sci. USA 2015, 112, E4919–E4928.
- Goldberg, E. Recognition, Attachment, and Injection. In Molecular Biology of Bacteriophage T4; Mathews, C.K., Kutter, E.M., Mosig, G., Berget, P.B., Eds.; American Society of Microbiology: Washington, DC, USA, 1983; pp. 32–39.
- Padilla-Sanchez, V. Structural Model of Bacteriophage T4. Wikij. Sci. 2021, 4, 5.
- Freifelder, D.E. Molecular Biology, 2nd ed.; Science Books International: Boston, MA, USA, 1983; p. 614.
- Cerritelli, M.E.; Wall, J.S.; Simon, M.N.; Conway, J.F.; Steven, A.C. Stoichiometry and domainal organization of the long tail-fiber of bacteriophage T4: A hinged viral adhesin. J. Mol. Biol. 1996, 260, 767–780.
- Leiman, P.G.; Arisaka, F.; van Raaij, M.J.; Kostyuchenko, V.A.; Aksyuk, A.A.; Kanamaru, S.; Rossmann, M.G. Morphogenesis of the T4 tail and tail fibers. Virol. J. 2010, 7, 355.
- Bartual, S.G.; Otero, J.M.; Garcia-Doval, C.; Llamas-Saiz, A.L.; Kahn, R.; Fox, G.C.; van Raaij, M.J. Structure of the bacteriophage T4 long tail fiber receptor-binding tip. Proc. Natl. Acad. Sci. USA 2010, 107, 20287–20292.
- Dunne, M.; Prokhorov, N.S.; Loessner, M.J.; Leiman, P.G. Reprogramming bacteriophage host range: Design principles and strategies for engineering receptor binding proteins. Curr. Opin. Biotechnol. 2021, 68, 272–281.
- King, J.; Laemmli, U.K. Polypeptides of the tail fibres of bacteriophage T4. J. Mol. Biol. 1971, 62, 465–477.
- Hashemolhosseini, S.; Stierhof, Y.D.; Hindennach, I.; Henning, U. Characterization of the helper proteins for the assembly of tail fibers of coliphages T4 and lambda. J. Bacteriol. 1996, 178, 6258–6265.
- Nobrega, F.L.; Vlot, M.; de Jonge, P.A.; Dreesens, L.L.; Beaumont, H.J.E.; Lavigne, R.; Dutilh, B.E.; Brouns, S.J.J. Targeting mechanisms of tailed bacteriophages. Nat. Rev. Microbiol. 2018, 16, 760–773.
- Islam, M.Z.; Fokine, A.; Mahalingam, M.; Zhang, Z.; Garcia-Doval, C.; van Raaij, M.J.; Rossmann, M.G.; Rao, V.B. Molecular anatomy of the receptor binding module of a bacteriophage long tail fiber. PLoS Pathog. 2019, 15, e1008193.
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410.
- Wang, C.; Tu, J.; Liu, J.; Molineux, I.J. Structural dynamics of bacteriophage P22 infection initiation revealed by cryo-electron tomography. Nat. Microbiol. 2019, 4, 1049–1056.
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. The bacteriophage t7 virion undergoes extensive structural remodeling during infection. Science 2013, 339, 576–579.
- North, O.I.; Sakai, K.; Yamashita, E.; Nakagawa, A.; Iwazaki, T.; Büttner, C.R.; Takeda, S.; Davidson, A.R. Phage tail fibre assembly proteins employ a modular structure to drive the correct folding of diverse fibres. Nat. Microbiol. 2019, 4, 1645–1653.
- Garcia-Doval, C.; van Raaij, M.J. Structure of the receptor-binding carboxy-terminal domain of bacteriophage T7 tail fibers. Proc. Natl. Acad. Sci. USA 2012, 109, 9390–9395.
- Subramanian, S.; John, A.D.; Kristin, N.P.; Sarah, M.D.; Rebecca, E.D. Host Range Expansion of Shigella Phage Sf6 Evolves through Point Mutations in the Tailspike. J. Virol. 2022, 96, e0092922.
- Šiborová, M.; Füzik, T.; Procházková, M.; Nováček, J.; Benešík, M.; Nilsson, A.S.; Plevka, P. Tail proteins of phage SU10 reorganize into the nozzle for genome delivery. Nat. Commun. 2022, 13, 5622.
- Linares, R.; Arnaud, C.-A.; Effantin, G.; Darnault, C.; Epalle, N.H.; Erba, E.B.; Schoehn, G.; Breyton, C. Structural basis of bacteriophage T5 infection trigger and E. coli cell wall perforation. bioRxiv 2022.
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide Endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700.
- Bertani, B.; Ruiz, N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018, 8.
- Washizaki, A.; Yonesaki, T.; Otsuka, Y. Characterization of the interactions between Escherichia coli receptors, LPS and OmpC, and bacteriophage T4 long tail fibers. Microbiologyopen 2016, 5, 1003–1015.
- Kalynych, S.; Morona, R.; Cygler, M. Progress in understanding the assembly process of bacterial O-antigen. FEMS Microbiol. Rev. 2014, 38, 1048–1065.
- Schnaitman, C.A.; Klena, J.D. Genetics of lipopolysaccharide biosynthesis in enteric bacteria. Microbiol. Rev. 1993, 57, 655–682.
- Moore, R.N.; Amor, K.; David, E.H.; Frirdich, E.; Ziebell, K.; Roger, P.J.; Whitfield, C. Distribution of Core Oligosaccharide Types in Lipopolysaccharides from Escherichia coli. Infect. Immun. 2000, 68, 1116–1124.
- Suga, A.; Kawaguchi, M.; Yonesaki, T.; Otsuka, Y. Manipulating Interactions between T4 Phage Long Tail Fibers and Escherichia coli Receptors. Appl. Environ. Microbiol. 2021, 87, e0042321.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82.
- Rosas, N.C.; Lithgow, T. Targeting bacterial outer-membrane remodelling to impact antimicrobial drug resistance. Trends Microbiol. 2022, 30, 544–552.
- Vergalli, J.; Bodrenko, I.V.; Masi, M.; Moynié, L.; Acosta-Gutiérrez, S.; Naismith, J.H.; Davin-Regli, A.; Ceccarelli, M.; van den Berg, B.; Winterhalter, M.; et al. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat. Rev. Microbiol. 2020, 18, 164–176.
- Rakhuba, D.V.; Kolomiets, E.I.; Dey, E.S.; Novik, G.I. Bacteriophage receptors, mechanisms of phage adsorption and penetration into host cell. Pol. J. Microbiol. 2010, 59, 145–155.
- Yu, S.L.; Ko, K.L.; Chen, C.S.; Chang, Y.C.; Syu, W.J. Characterization of the distal tail fiber locus and determination of the receptor for phage AR1, which specifically infects Escherichia coli O157:H7. J. Bacteriol. 2000, 182, 5962–5968.
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327.
- Altamirano, F.G.; Forsyth, J.H.; Patwa, R.; Kostoulias, X.; Trim, M.; Subedi, D.; Archer, S.K.; Morris, F.C.; Oliveira, C.; Kielty, L.; et al. Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 2021, 6, 157–161.
- Wang, X.; Loh, B.; Altamirano, F.G.; Yu, Y.; Hua, X.; Leptihn, S. Colistin-phage combinations decrease antibiotic resistance in Acinetobacter baumannii via changes in envelope architecture. Emerg. Microbes Infect. 2021, 10, 2205–2219.
- Goodsell, D.S. Escherichia coli. Biochem. Mol. Biol. Educ. 2009, 37, 325–332.
- Rossmann, M.G.; Mesyanzhinov, V.V.; Arisaka, F.; Leiman, P.G. The bacteriophage T4 DNA injection machine. Curr. Opin. Struct. Biol. 2004, 14, 171–180.
- Hampton, H.G.; Watson, B.N.J.; Fineran, P.C. The arms race between bacteria and their phage foes. Nature 2020, 577, 327–336.
- Dy, R.L.; Richter, C.; Salmond, G.P.C.; Fineran, P.C. Remarkable Mechanisms in Microbes to Resist Phage Infections. Annu. Rev. Virol. 2014, 1, 307–331.
- Habusha, M.; Tzipilevich, E.; Fiyaksel, O.; Ben-Yehuda, S. A mutant bacteriophage evolved to infect resistant bacteria gained a broader host range. Mol. Microbiol. 2019, 111, 1463–1475.
- Pawluk, A.; Davidson, A.R.; Maxwell, K.L. Anti-CRISPR: Discovery, mechanism and function. Nat. Rev. Microbiol. 2018, 16, 12–17.
- Srikant, S.; Guegler, C.K.; Laub, M.T. The evolution of a counter-defense mechanism in a virus constrains its host range. eLife 2022, 11, e79549.
- Molineux, I.J. The T7 group. Bacteriophages 2005, 2, 277–301.
- Burrowes, B.H.; Molineux, I.J.; Fralick, J.A. Directed in vitro evolution of therapeutic bacteriophages: The Appelmans protocol. Viruses 2019, 11, 241.
- Yoichi, M.; Abe, M.; Miyanaga, K.; Unno, H.; Tanji, Y. Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157: H7. J. Biotechnol. 2005, 115, 101–107.
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst. 2015, 1, 187–196.
- Yosef, I.; Goren, M.G.; Globus, R.; Molshanski-Mor, S.; Qimron, U. Extending the host range of bacteriophage particles for DNA transduction. Mol. Cell 2017, 66, 721–728.e3.
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.D.T.; de la Fuente-Nunez, C.; Lu, T.K. Engineering phage host-range and suppressing bacterial resistance through phage tail fiber mutagenesis. Cell 2019, 179, 459–469.e9.
- Huss, P.; Meger, A.; Leander, M.; Nishikawa, K.; Raman, S. Mapping the functional landscape of the receptor binding domain of T7 bacteriophage by deep mutational scanning. eLife 2021, 10, e63775.
- Ahlgren, N.A.; Ren, J.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. Alignment-free oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically-derived viral sequences. Nucleic Acids Res. 2017, 45, 39–53.
- Versoza, C.J.; Pfeifer, S.P. Computational Prediction of Bacteriophage Host Ranges. Microorganisms 2022, 10, 149.
- Meng, C.; Zhang, J.; Ye, X.; Guo, F.; Zou, Q. Review and comparative analysis of machine learning-based phage virion protein identification methods. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2020, 1868, 140406.
- Cantu, V.A.; Salamon, P.; Seguritan, V.; Redfield, J.; Salamon, D.; Edwards, R.A.; Segall, A.M. PhANNs, a fast and accurate tool and web server to classify phage structural proteins. PLoS Comput. Biol. 2020, 16, e1007845.
- Dunne, M.; Rupf, B.; Tala, M.; Qabrati, X.; Ernst, P.; Shen, Y.; Sumrall, E.; Heeb, L.; Plückthun, A.; Loessner, M.J.; et al. Reprogramming Bacteriophage Host Range through Structure-Guided Design of Chimeric Receptor Binding Proteins. Cell Rep. 2019, 29, 1336–1350.e4.
- Boeckaerts, D.; Stock, M.; Criel, B.; Gerstmans, H.; De Baets, B.; Briers, Y. Predicting bacteriophage hosts based on sequences of annotated receptor-binding proteins. Sci. Rep. 2021, 11, 1467.
- Ataee, S.; Brochet, X.; Peña-Reyes, C.A. Bacteriophage Genetic Edition using LSTM. Front. Bioinform. 2022, 73, 932319.