Erythromycin (ERY) is a macrolide compound with a broad antimicrobial spectrum which is currently being used to treat a large number of bacterial infections affecting the skin, respiratory tract, intestines, bones and other systems, proving great value from a clinical point of view. Despite this major advantage, ERY has low water solubility and is not stable under acidic conditions which leads to a limited efficacy and bioavailability. Apart from this, higher doses promote drug resistance and undesirable effects. In order to overcome these disadvantages, during the past decades, a large variety of ERY formulations, including nanoparticles, have emerged. This work presents the preparation and performances reported for ERY vesicles, such as liposomes, ethosomes, niosomes, micelles, cubosomes and solid lipid nano(micro) particles.
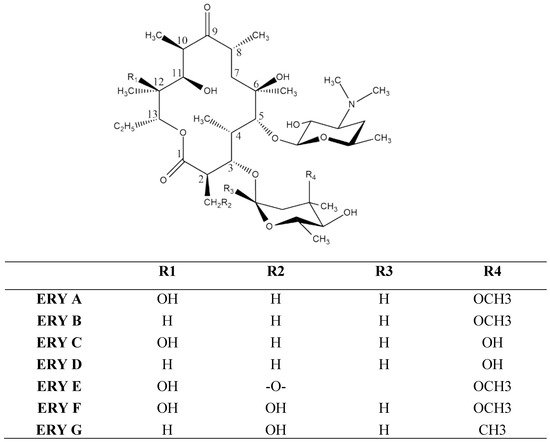
- erythromycin
- liposomes
- solid–lipid nanoparticles
- ethosomes
- niosomes
- micelles
- cubosomes
1. Introduction
2. Vesicles
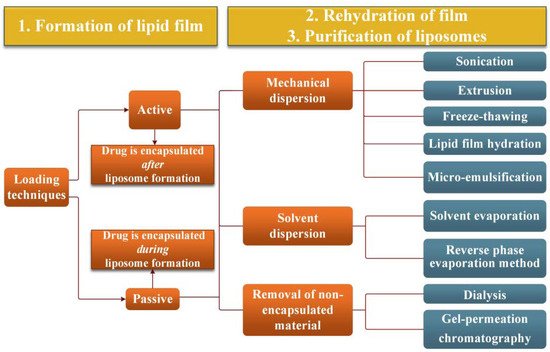
The literature reveals a large variety of vesicular systems used to deliver drugs, such as lipid particles, liposomes, niosomes, sphingosomes, pharmacosomes, transferosomes, polymeric micelles, etc.[24]. Generally speaking, vesicular carriers are formulated with lipid materials for the purpose of overcoming challenges of drug delivery and improving therapeutic outcomes. Encapsulation of both hydrophilic and lipophilic drugs, enhancement of bioavailability, increasing the drug’s circulation time, resolving toxicity issues and lowering therapeutic dosages are some of the major benefits of lipid-based drug delivery. In the following, ERY vesicle formulations developed over a long time; their preparation and performances will be presented.2.1. Liposomes
Liposomes are sphere-shaped vesicles of small diameter, ranging from a few nanometres to several micrometres, consisting of aqueous compartments surrounded by one or more lipid bilayers (Figure 2)[25].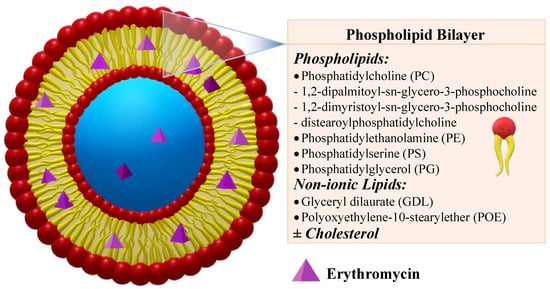
2.2. Ethosomes
To improve drug delivery performance of liposomes, soft vesicles, mainly containing molecules with the capacity to induce more fluidity into the phospholipid bilayers, were proposed. When ethanol (20–50%) is entrapped into liposomes, the soft vesicles are called ethosomes[53]. Compared to liposomes, which remain confined to the upper stratum corneum of the skin, ethosomes exhibit a higher potential to transport active therapeutic agents into deeper skin layers, due to the permeation enhancing capacity of ethanol[54][55]. It was stated that ethanol can fluidize both the vesicles and the stratum corneum lipids, making the skin more penetrable, while the vesicles become more flexible. ERY ethosomes were prepared in a well-sealed container by dissolving phospholipon 90 and ERY in ethanol, followed by the slow addition of distilled water under constant stirring at 30 °C[56]. They displayed a spherical unilamelar configuration with low size (116 and 123 nm), EE of ~78%, improved bilayer fluidity and excellent shelf-life, their size and configuration being almost unaffected after one year of storage under ambiental conditions (Figure 4). The investigation of the antibacterial properties against S. aureus and B. subtills showed improved activity when compared to standard hydroethanolic solutions, with remarkable reduction of the MIC of 2.5 times, against a clinically isolated S. aureus strain with known resistance to ERY. These results indicated an improvement of ERY delivery from the ethosomes to the bacteria, which was attributed to an enhanced permeability of microorganisms to ethosomal ERY, suggesting the necessity of administrating lower doses of the drug. Moreover, this researchtudy suggested that ethosomes are a good approach to overcome bacterial resistance to antibiotics.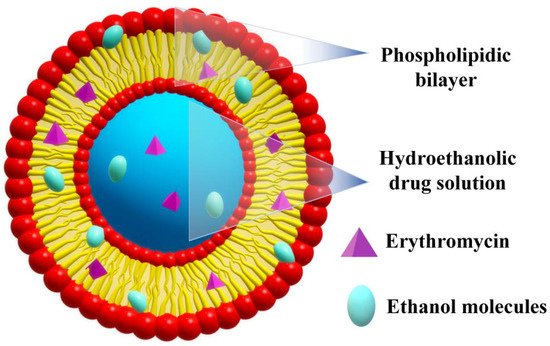
2.3. Niosomes
Niosomes are slightly modified version of liposomes, in which the lipid is replaced by non-ionic surfactants (Figure 5)[58]. They are biodegradable, relatively nontoxic, and more stable, yet less expensive compared to liposomes. Drug-loaded niosomes show interactions with the epidermal tissue without presenting an immediate or strong systemic action, thus being preferred for dermal applications.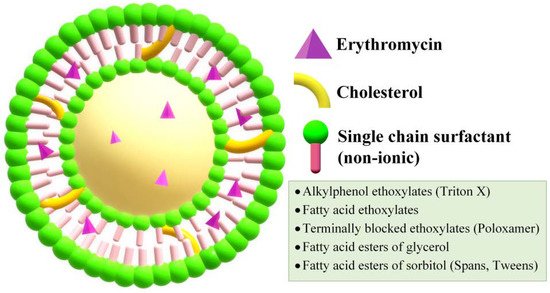
2.4. Micelles
Micelles are obtained by similar preparation methods and composition as niosomes, with the difference that cholesterol is absent (Figure 7). Liu et al. developed a micelle formulation encapsulating ERY with the use of Pluronic F-127 nonionic surfactant[62]. The obtained micelles had an EE of 28.3%, and they displayed a spherical morphology with an approximate diameter of 193 nm. In vitro release studies concluded that over 90% of the drug was released over 8 h, but the antibacterial activity tested on strains of S. aureus and Escherichia coli (E. coli) revealed MIC values five times higher than those of simple ERY.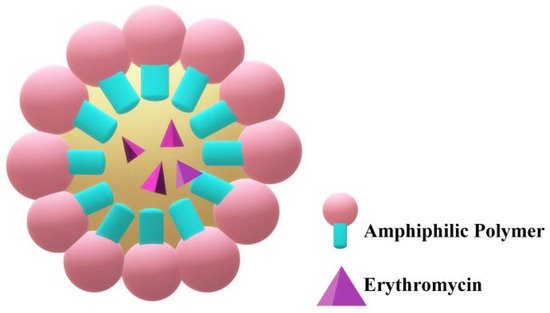
2.5. Cubosomes
Cubosomes are discrete, sub-micron nanostructured particles comprising a bicontinuous cubic liquid crystalline phase, possessing a good ability to penetrate the skin even through the pilosebaceous units[66]. They are obtained by the self-assembly of certain surfactants and lipids in water, enabling the entrapment of hydrophobic and hydrophilic drugs due to their bicontinuous nature (Figure 8).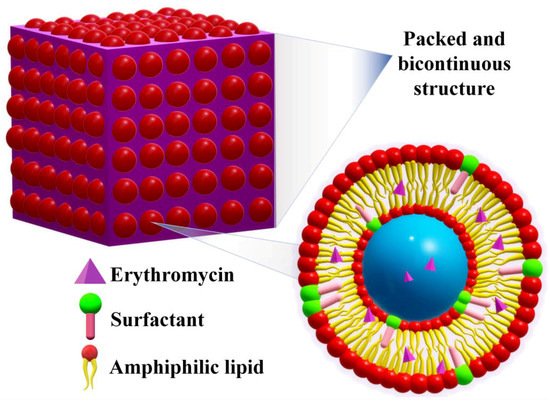
2.6. Solid Lipid Nano(Micro)Particles
Literature data mentioned the preparation of solid lipid nanoparticles (SLN) and solid lipid microparticles (SLM), the classification being created by their dimension. Nevertheless, the similarity of the composition of SLNs and SLMs induce similar physicochemical properties, the main distinction being their application fields and administration routes that are dictated by their different size[68]. Furthermore, the use of the term “nano” has varied along the course of time[69][70]. Therefore, in this section, solid lipid nano(micro)particles shall be presented as they were reported, rather than their differentiation based strictly on size criteria. The distinct characteristic of the solid lipid nano(micro)particles is the presence of biodegradable lipid-based components found in a solid state at physiological temperatures (Figure 9). Some examples of such components are hard fats such as glycerol (mono-, di-, and/or triglycerides), complex glyceride mixtures, fatty acids, steroids or even waxes[71]. One of the obvious advantages of solid lipid particles is that the lipid matrix is composed of physiological lipids, thus significantly decreasing the risk of toxicity[72]. Therefore, the mentioned drug delivery system was promoted as a safer option, in addition to the higher stability of the solid lipid matrix compared to other nano(micro)particles, and the cheaper production costs than those of phospholipid-based liposomes developed thus far[73]. They offer great potential for the administration of active molecules by any route, including tablets and suppositories.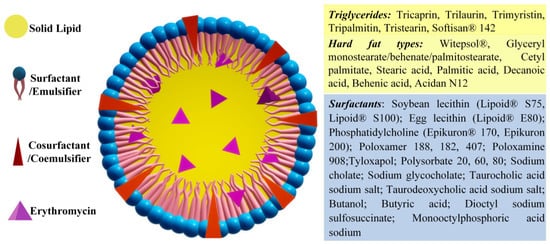
2.6.1. Solid Lipid Nanoparticles
An exhaustive study regarding the preparation and composition optimization of ERY loaded solid lipid nanoparticles (ERY-SLN) was realized by using the hot homogenization method choosing glycerol monostearate to act as the lipid phase, Poloxamer 188 as surfactant and soya lecithin as a cosurfactant[74]. Even though no other component was mentioned, the DSC study makes reference to the presence of mannitol as a cryoprotectant. Considering that the surfactant concentration can shape the properties of SLNs, different compositions were investigated, and it was found that 2% (w/v) Poloxamer 188 is optimal to reach the smallest particle size of 138 nm, with the highest EE and DL (84.5% and 28.1%, respectively). Studying the effect of different variables on SLN properties via a miscellaneous design–response surface methodology, the researcheuthors concluded that the increase in lipid concentration produced a decrease in the mean particle size but also in the entrapment efficiency. Therefore, a minimum particle size of 153 nm and maximum EE of 88.4% were reached when using a lipid concentration of 15 mg/mL and surfactant/cosurfactant ratio of 1.07/1. The ERY-SLN presented negative Zeta potential values up to −19 mV, reflecting an excellent stability. No significant alteration of the particle size and EE was observed for the SLN dispersions stored at 4 or 25 °C, over a three-month period. The release profile, which was investigated in phosphate buffer media, showed a prolonged release during 24 h, up to 66.26 ± 2.83%. Pursuing the preparation of a topical formulation aimed at the treatment of acne, Dhillon et al. investigated the development and optimization of ERY-loaded SLNs. The study focused on selecting the best preparation method, lipid type, drug/lipid ratio, stirring, and sonication time[75]. The findings led to the conclusion that the microencapsulation technique was superior to the solvent evaporation and the solvent emulsification diffusion ones, because it showed better results in terms of particle size, particle shape and drug entrapment. The screening of three lipids, stearic acid, glycerol monostearate and Compatriol 888, revealed that the use of glycerol monostearate significantly decreased the particle size into the nanometric range and favoured the highest ERY entrapment. Furthermore, it was discovered that an ERY/glycerol monostearate ratio of ½ was optimal for a maximum encapsulation of 78% into nanoparticles of 176 nm. The researcheuthors also reported that a 0.5% concentration of surfactant (Poloxamer 188) was enough to preserve the size of the nanoparticles and to ensure a high Zeta potential, required for particle stability. An increase in stirring time (from 30 to 60 min) produced a dramatic decrease in particle size (from 528 to 176 nm) and higher entrapment degree values, which reached 78.59%. A sonication time of 20 min was optimal to reduce the particle size. The average particle size, PDI, Zeta-potential, EE and LD of the optimized SLNs were found to be ~176 nm, 0.275, −34 mV, 73.56% and 69.74% respectively. They showed a prolonged release, reaching around 85% cumulative ERY release after 12 h and 93% after 24 h. Targeting the improvement of ERY’s penetration into bacterial cells, cationic SLNs were designed using a cationic lipid, quaternary ammonium salt and didecyldimethylammonium bromide[76]. SLNs with mean diameter from 250 to 400 nm were prepared using the solvent injection method. The cationic lipid induced positive Zeta potential and great stability over time, with no change in the morphologic parameters being recorded after one year of storage. As proof of concept, a higher antimicrobial activity of SLNs compared to free ERY was evidenced, and the MIC decreased along with the increase in cationic lipid content, proving its synergic antibacterial activity.2.6.2. Solid Lipid Microparticles
ERY-loaded SLMs were prepared for oral delivery, using a binary mixture of ½ ratio solid lipids (Softisan154 and Phospholipon90H or Softisan154 and beeswax) forming a solidified reverse micellar solution (SRMS) matrix. PVA, sorbitol and sorbic acid were also used as surfactants[77]. Priorly, the SRMS matrix was prepared by fusion, and the SLMs were prepared by hot homogenization method, varying the ERY concentration in order to obtain SLMs with different drug contents. The DSC investigation suggested the occurrence of interactions between the two lipids, leading to a less crystalline morphology, envisioned to have a higher ability to retain ERY over time compared to a more crystalline matrix, which is prone to a faster drug expulsion. The SLMs showed a spherical morphology, with a smooth and non-porous surface, while the diameters increased proportionally from 13 to 17 µm along with the entrapped ERY amount, with no significant distinctions between those containing different lipid mixtures. The nature of the lipids influenced the EE and LD, i.e., the SLMs composed of Phospholipon90H displayed higher EE values, ranging from 89 to 95%, while those containing beeswax showed variations of the EE from 78 to 89%. They were stable over the span of 4 weeks; however, an aggregation tendency towards the end was observed, while a slight pH shift towards acidity suggested the degradation of the lipid component to fatty acids. The in vitro release studies revealed that all ERY-loaded SLMs possessed higher prolonged release properties and no burst effect or dose dumping, compared to the reference ERY-tablets. This suggests a reduction in the daily number of administrations (from 3–4 to 1–2) needed to reach and maintain the optimal plasma concentration. The release profile was dose dependent and was influenced by the nature of the lipid; SLMs containing Phospholipon90H exhibited a maximum release ranging from 90 to 75%, while those containing bee waxes ranged from 79 to 61%. The antimicrobial activity tests against S. aureus, S. paratyphii, P. aeruginosa, and E. coli confirmed a time- and dose-dependent anti-bacterial activity, higher for those containing Phospholipon90H compared to bee waxes and simple ERY. The findings suggest that the formulation of ERY as SLMs potentiates its activity, possibly due to the fusion between the lipid component and the bacterial cell membranes, promoting an easier penetration of the antibiotic. Pharmacokinetical studies on rats showed a steady and slow decrease in the drug concentrations or a gradual clearance, reaching a T1/2 value for the SLMs of 8 h, compared to a T1/2 of 4.5 h in the case of the neat ERY. ERY-loaded SLMs were also prepared using stearic acid as the lipid component and Tween 80 as surfactant, via hot homogenization method[78]. By varying the lipid/surfactant ratio, the average hydrodynamic diameters ranged from 2.28 to 8.15 μm, the lowest particle size being reached for a lipid/surfactant ratio of 1 g/1 mL and 1 g/2 mL. The melting temperature of ERY was clearly identified in the DSC curves, indicating that it was dispersed as large particles into the lipid, attributed to the ERY insolubility into the lipid matrix. The microparticles showed much higher in vitro antimicrobial activity compared to neat ERY; however, those with the lowest MIC values were large in size (6.5 μm), at risk of not being able to pass into the lymphatic circulation[79]. Furthermore, the microparticles containing higher amounts of surfactant and thus more stable in acidic environment, were selected for in vivo tests on model infected animals. It was proven that the administration of equal amounts of SLM and neat ERY led to almost similar bacteremia reduction (75% vs. 72% during day 3, and 95% vs. 94% during day 7), despite the small percentage of ERY entrapped into SLMs (21%). This suggested a significant increase in the antimicrobial activity of ERY when encapsulated into SLMs. Further, only low fluctuations in plasma concentrations were recorded in the case of SLMs, indicating a potential to reduce major side effects.References
- Bunch, R.L.; Mcguire, J.M. Erythromycin, Its Salts, and Method of Preparation. U.S. Patent 2,653,899, 29 September 1952.
- P. Dehouck; Y. Vander Heyden; J. Smeyers-Verbeke; D.L. Massart; R.D. Marini; P. Chiap; Ph. Hubert; J. Crommen; W. Van de Wauw; J. De Beer; et al.R. CoxG. MathieuJ.C. ReepmeyerB. VoigtO. EstevenonA. NicolasA. Van SchepdaelE. AdamsJ. Hoogmartens Interlaboratory study of a liquid chromatography method for erythromycin: determination of uncertainty. Journal of Chromatography A 2003, 1010, 63-74, 10.1016/s0021-9673(03)01023-9.
- P. Dehouck; Y. Vander Heyden; J. Smeyers-Verbeke; D.L. Massart; R.D. Marini; P. Chiap; Ph. Hubert; J. Crommen; W. Van de Wauw; J. De Beer; et al.R. CoxG. MathieuJ.C. ReepmeyerB. VoigtO. EstevenonA. NicolasA. Van SchepdaelE. AdamsJ. Hoogmartens Interlaboratory study of a liquid chromatography method for erythromycin: determination of uncertainty. Journal of Chromatography A 2003, 1010, 63-74, 10.1016/s0021-9673(03)01023-9.
- F R Heilman; W E Herrell; W E Wellman; J E Geraci; Some laboratory and clinical observations on a new antibiotic, erythromycin (ilotycin).. Proceedings of the staff meetings. Mayo Clinic 1952, 27, 285–304.
- F R Heilman; W E Herrell; W E Wellman; J E Geraci; Some laboratory and clinical observations on a new antibiotic, erythromycin (ilotycin).. Proceedings of the staff meetings. Mayo Clinic 1952, 27, 285-304.
- Jeffrey C. Hoyt; Macrolide antibiotics and pulmonary inflammation. FEMS Microbiology Letters 2001, 205, 1-7, 10.1016/s0378-1097(01)00443-8.
- Stan Block; James Hedrick; Margaret R. Hammerschlag; Gail H. Cassell; J. Carl Craft; Mycoplasma pneumoniae and Chlamydia pneumoniae in pediatric community-acquired pneumonia. Pediatric Infectious Disease Journal 1995, 14, 471-477, 10.1097/00006454-199506000-00002.
- Horwitz, M.A.; Silverstein, S.C. Intracellular Multiplication of Legionnaires’ Disease Bacteria (Legionella Pneumophila) in Human Monocytes Is Reversibly Inhibited by Erythromycin and Rifampin. J. Clin. Investig. 1983, 71, 15–26.
- Rachel Kneen; Pham Ngoc Giao; Tom Solomon; Tran Thi My Van; Nguyen Thi Tuyet Hoa; Tran Buu Long; John Wain; Nicholas P. J. Day; Tran Tinh Hien; Christopher M. Parry; et al.Nicholas J. White Penicillin vs. Erythromycin in the Treatment of Diphtheria. Clinical Infectious Diseases 1998, 27, 845-850, 10.1086/514959.
- J. M. Rolain; P. Brouqui; J. E. Koehler; C. Maguina; M. J. Dolan; D. Raoult; Recommendations for Treatment of Human Infections Caused by Bartonella Species. Antimicrobial Agents and Chemotherapy 2004, 48, 1921-1933, 10.1128/aac.48.6.1921-1933.2004.
- Eduardo Salazar-Lindo; R. Bradley Sack; Elsa Chea-Woo; Bradford A. Kay; Zolia A. Piscoya; Raul Leon-Barua; August Yi; Early treatment with erythromycin of Campylobacter jejuni-associated dysentery in children. The Journal of Pediatrics 1986, 109, 355-360, 10.1016/s0022-3476(86)80404-8.
- Leslie Slaney; Hilary Chubb; Allan Ronald; Robert Brunham; In-vitro activity of azithromycin, erythromycin, ciprofloxacin and norfloxacin against Neisseria gonorrhoeae, Haemophilus ducreyi, and Chlamydia trachomatis. Journal of Antimicrobial Chemotherapy 1990, 25, 1-5, 10.1093/jac/25.suppl_a.1.
- Zheng Xu; Zengguo Wang; Yang Luan; Yarong Li; Xiaoguai Liu; Xiaokang Peng; Sophie Octavia; Michael Payne; Ruiting Lan; Genomic epidemiology of erythromycin-resistant Bordetella pertussis in China. Emerging Microbes & Infections 2019, 8, 461-470, 10.1080/22221751.2019.1587315.
- G.F. Webster; K.J. McGINLEY; J.J. Leyden; Inhibition of lipase production in Propionibacterium acnes by sub-minimal-inhibitory concentrations of tetracycline and erythromycin. British Journal of Dermatology 1981, 104, 453-457, 10.1111/j.1365-2133.1981.tb15317.x.
- Hikaru Tamura; Tomoki Maekawa; Hisanori Domon; Takumi Hiyoshi; Satoru Hirayama; Toshihito Isono; Karin Sasagawa; Daisuke Yonezawa; Naoki Takahashi; Masataka Oda; et al.Takeyasu MaedaKoichi TabetaYutaka Terao Effects of Erythromycin on Osteoclasts and Bone Resorption via DEL-1 Induction in Mice. Antibiotics 2021, 10, 312, 10.3390/antibiotics10030312.
- Samit Kumar Nandi; Prasenjit Mukherjee; Subhasis Roy; Biswanath Kundu; Dipak Kumar De; Debabrata Basu; Local antibiotic delivery systems for the treatment of osteomyelitis – A review. Materials Science and Engineering: C 2009, 29, 2478-2485, 10.1016/j.msec.2009.07.014.
- F H Weber; R D Richards; R W McCallum; Erythromycin: a motilin agonist and gastrointestinal prokinetic agent.. American Journal of Gastroenterology 1993, 88, 485–490.
- G J Sanger; Y Wang; A Hobson; J Broad; Motilin: towards a new understanding of the gastrointestinal neuropharmacology and therapeutic use of motilin receptor agonists. British Journal of Pharmacology 2013, 170, 1323-1332, 10.1111/bph.12075.
- Yang Fan; Wu Shan-Guang; Pan Yu-Fang; Song Feng-Lan; Li Tao; Preparation and characteristics of erythromycin microspheres for lung targeting. Drug Development and Industrial Pharmacy 2009, 35, 639-645, 10.1080/03639040802512243.
- Zhanzhong Wang; Jingkang Wang; And Meijing Zhang; Leping Dang; Solubility of Erythromycin A Dihydrate in Different Pure Solvents and Acetone + Water Binary Mixtures between 293 K and 323 K. Journal of Chemical & Engineering Data 2006, 51, 1062-1065, 10.1021/je0505265.
- Barry L. Carter; Jerold C. Woodhead; Katherine J. Cole; Gary Milavetz; Gastrointestinal Side Effects with Erythromycin Preparations. Drug Intelligence & Clinical Pharmacy 1987, 21, 734-738, 10.1177/106002808702100914.
- Dave Baguley; Emma Lim; Amanda Bevan; Ann Pallet; Saul Faust; Prescribing for children – taste and palatability affect adherence to antibiotics: a review. Archives of Disease in Childhood 2011, 97, 293-297, 10.1136/archdischild-2011-300909.
- Isadore Kanfer; Michael F Skinner; Roderick B Walker; Analysis of macrolide antibiotics. Journal of Chromatography A 1998, 812, 255-286, 10.1016/s0021-9673(98)00276-3.
- Jadhav, S.M.; Morey, P.; Karpe, M.M.; Kadam, V. Novel Vesicular System: An Overview. J. Appl. Pharm. Sci. 2012, 2, 193–202.
- Roxana-Maria Amarandi; Alina Ibanescu; Eugen Carasevici; Luminita Marin; Brindusa Dragoi; Liposomal-Based Formulations: A Path from Basic Research to Temozolomide Delivery Inside Glioblastoma Tissue. Pharmaceutics 2022, 14, 308, 10.3390/pharmaceutics14020308.
- Maria-Lucia Briuglia; Carlo Maria Rotella; Amber McFarlane; Dimitrios A. Lamprou; Influence of cholesterol on liposome stability and on in vitro drug release. Drug Delivery and Translational Research 2015, 5, 231-242, 10.1007/s13346-015-0220-8.
- W.W. Sułkowski; Danuta Pentak; K. Nowak; A. Sułkowska; The influence of temperature, cholesterol content and pH on liposome stability. Journal of Molecular Structure 2005, 744-747, 737-747, 10.1016/j.molstruc.2004.11.075.
- Sriram Vemuri; C.T Rhodes; Preparation and characterization of liposomes as therapeutic delivery systems: a review. Pharmaceutica Acta Helvetiae 1995, 70, 95-111, 10.1016/0031-6865(95)00010-7.
- Theresa M. Allen; Pieter R. Cullis; Liposomal drug delivery systems: From concept to clinical applications. Advanced Drug Delivery Reviews 2013, 65, 36-48, 10.1016/j.addr.2012.09.037.
- A.D. Bangham; M.M. Standish; J.C. Watkins; Diffusion of univalent ions across the lamellae of swollen phospholipids. Journal of Molecular Biology 1965, 13, 238-252, 10.1016/s0022-2836(65)80093-6.
- Omid Rajabi; Pouran Layegh; Sara Hashemzadeh; Mohsen Khoddami; Topical liposomal azithromycin in the treatment of acute cutaneous leishmaniasis. Dermatologic Therapy 2016, 29, 358-363, 10.1111/dth.12357.
- Bahareh Abtahi-Naeini; Sajjad Hadian; Fatemeh Sokhanvari; Amirali Hariri; Jaleh Varshosaz; Zabihollah Shahmoradi; Awat Feizi; Farzin Khorvash; Atousa Hakamifard; Effect of Adjunctive Topical Liposomal Azithromycin on Systemic Azithromycin on Old World Cutaneous Leishmaniasis: A Pilot Clinical Study. null 2021, 20, 383-389, 10.22037/IJPR.2020.113710.14445.
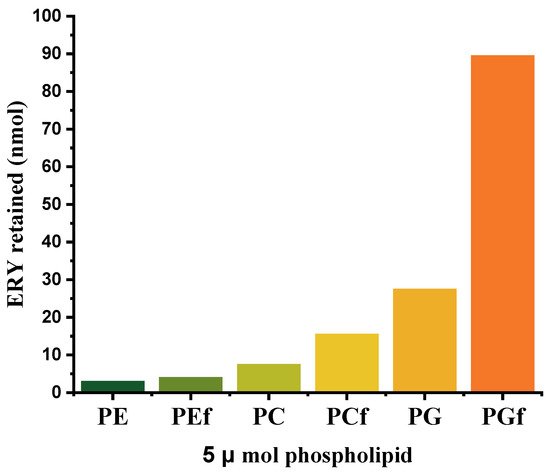
- L. Stuhne-Sekalec; N. Z. Stanacev; S. Djokic; Liposomes as carriers of macrolides: Preferential association of erythromycin A and azithromycin with liposomes of phosphatidylglycerol containing unsaturated fatty acid(s). Journal of Microencapsulation 1991, 8, 171-183, 10.3109/02652049109071486.
- Clement Mugabe; Ali O. Azghani; Abdelwahab Omri; Preparation and characterization of dehydration–rehydration vesicles loaded with aminoglycoside and macrolide antibiotics. International Journal of Pharmaceutics 2006, 307, 244-250, 10.1016/j.ijpharm.2005.10.005.
- Clement Mugabe; Ali O. Azghani; Abdelwahab Omri; Liposome-mediated gentamicin delivery: development and activity against resistant strains of Pseudomonas aeruginosa isolated from cystic fibrosis patients. Journal of Antimicrobial Chemotherapy 2005, 55, 269-271, 10.1093/jac/dkh518.
- Shrenik P. Shah; Ambikanandan Misra; Liposomal amikacin dry powder inhaler: Effect of fines on in vitro performance. AAPS PharmSciTech 2004, 5, 107-113, 10.1208/pt050465.
- Douweh Gbian; Abdelwahab Omri; The Impact of an Efflux Pump Inhibitor on the Activity of Free and Liposomal Antibiotics against Pseudomonas aeruginosa. Pharmaceutics 2021, 13, 577, 10.3390/pharmaceutics13040577.
- Frédérick de Meyer; Berend Smit; Effect of cholesterol on the structure of a phospholipid bilayer. Proceedings of the National Academy of Sciences 2009, 106, 3654-3658, 10.1073/pnas.0809959106.
- Huangliang Zheng; Hai Tao; Jinzhao Wan; Kei Yan Lee; Zhanying Zheng; Sharon Shui Yee Leung; Preparation of Drug-Loaded Liposomes with Multi-Inlet Vortex Mixers. Pharmaceutics 2022, 14, 1223, 10.3390/pharmaceutics14061223.
- Abolfazl Akbarzadeh; Rogaie Rezaei-Sadabady; Soodabeh Davaran; Sang Woo Joo; Nosratollah Zarghami; Younes Hanifehpour; Mohammad Samiei; Mohammad Kouhi; Kazem Nejati-Koshki; Liposome: classification, preparation, and applications. Nanoscale Research Letters 2013, 8, 102-102, 10.1186/1556-276x-8-102.
- Shyamala C. Jayaraman; C. Ramachandran; Norman Weiner; Topical Delivery of Erythromycin from Various Formulations: An in Vivo Hairless Mouse Study. Journal of Pharmaceutical Sciences 1996, 85, 1082-1084, 10.1021/js960040u.
- T J. Majack; M B. Rust; K C. Massee; . G. W. Kissil; R W. Hardy; M E. Peterson; Bioencapsulation of erythromycin using adult brine shrimp, Artemia franciscana (Latreille). Journal of Fish Diseases 2000, 23, 71-76, 10.1046/j.1365-2761.2000.00210.x.
- Loredana Nicoleta Hilițanu; Liliana Mititelu-Tarțău; Grațiela Eliza Popa; Beatrice Rozalina Buca; Liliana Lăcrămioara Pavel; Ana-Maria Pelin; Andreea-Daniela Meca; Maria Bogdan; Daniela Angelica Pricop; The Analysis of Chitosan-Coated Nanovesicles Containing Erythromycin—Characterization and Biocompatibility in Mice. Antibiotics 2021, 10, 1471, 10.3390/antibiotics10121471.
- Kulkarni, P.; Yadav, J.; Vaidya, K. Liposomes: A Novel Drug Delivery System. International Journal of Current Pharmaceutical Research. 2011, 3, 10–18.
- Nina Filipczak; Jiayi Pan; Satya Siva Kishan Yalamarty; Vladimir P. Torchilin; Recent advancements in liposome technology. Advanced Drug Delivery Reviews 2020, 156, 4-22, 10.1016/j.addr.2020.06.022.
- Songhee Jeong; Jonghwan Lee; Byeong Nam Im; Hyung Park; Kun Na; Combined photodynamic and antibiotic therapy for skin disorder via lipase-sensitive liposomes with enhanced antimicrobial performance. Biomaterials 2017, 141, 243-250, 10.1016/j.biomaterials.2017.07.009.
- Hyejin Park; Hyung Park; Kun Na; Dual Propionibacterium acnes therapy using skin penetration-enhanced liposomes loaded with a photosensitizer and an antibiotic. Journal of Porphyrins and Phthalocyanines 2015, 19, 956-966, 10.1142/s1088424615500686.
- Abolfazl Akbarzadeh; Mohammad Samiei; Soodabeh Davaran; Magnetic nanoparticles: preparation, physical properties, and applications in biomedicine. Nanoscale Research Letters 2012, 7, 144-144, 10.1186/1556-276x-7-144.
- Bassant M. Salah; Mai Rady; Mohammad Abdel-Halim; Heba M. Fahmy; Nermeen S. El-Din; Mohamed H. Gaber; Alternating Magnetic Field Induced Membrane Permeability in Erythromycin Magneto-Liposomes A Potential Solution to Antibiotic Resistance. Biophysics 2021, 66, 264-272, 10.1134/s0006350921020196.
- Joan Estelrich; Elvira Escribano; Josep Queralt; Maria Antònia Busquets; Iron Oxide Nanoparticles for Magnetically-Guided and Magnetically-Responsive Drug Delivery. International Journal of Molecular Sciences 2015, 16, 8070-8101, 10.3390/ijms16048070.
- Nicusor Fifere; Anton Airinei; Daniel Timpu; Aurelian Rotaru; Liviu Sacarescu; Laura Ursu; New insights into structural and magnetic properties of Ce doped ZnO nanoparticles. Journal of Alloys and Compounds 2018, 757, 60-69, 10.1016/j.jallcom.2018.05.031.
- J.-P. Montenez; Françoise Van Bambeke; J. Piret; R. Brasseur; P.M. Tulkens; M.-P. Mingeot-Leclercq; Interactions of Macrolide Antibiotics (Erythromycin A, Roxithromycin, Erythromycylamine [Dirithromycin], and Azithromycin) with Phospholipids: Computer-Aided Conformational Analysis and Studies on Acellular and Cell Culture Models. Toxicology and Applied Pharmacology 1999, 156, 129-140, 10.1006/taap.1999.8632.
- Ibrahim M. Abdulbaqi; Yusrida Darwis; Nurzalina Abdul Karim Khan; Reem Abou Assi; Arshad Ali Khan; Ethosomal nanocarriers: the impact of constituents and formulation techniques on ethosomal properties, in vivo studies, and clinical trials. International Journal of Nanomedicine 2016, ume 11, 2279-2304, 10.2147/ijn.s105016.
- Elka Touitou; Hiba Natsheh; Topical Administration of Drugs Incorporated in Carriers Containing Phospholipid Soft Vesicles for the Treatment of Skin Medical Conditions. Pharmaceutics 2021, 13, 2129, 10.3390/pharmaceutics13122129.
- Hiba Natsheh; Elka Touitou; Phospholipid Vesicles for Dermal/Transdermal and Nasal Administration of Active Molecules: The Effect of Surfactants and Alcohols on the Fluidity of Their Lipid Bilayers and Penetration Enhancement Properties. Molecules 2020, 25, 2959, 10.3390/molecules25132959.
- Biana Godin; Elka Touitou; Erythromycin Ethosomal Systems: Physicochemical Characterization and Enhanced Antibacterial Activity. Current Drug Delivery 2005, 2, 269-275, 10.2174/1567201054367931.
- B. Godin; E. Touitou; E. Rubinstein; A. Athamna; M. Athamna; A new approach for treatment of deep skin infections by an ethosomal antibiotic preparation: an in vivo study. Journal of Antimicrobial Chemotherapy 2005, 55, 989-994, 10.1093/jac/dki125.
- Ketousetuo Kuotsu; Kazi Masud Karim; Asim Sattwa Mandal; Nikhil Biswas; Arijit Guha; Sugata Chatterjee; Mamata Behera; Niosome: A future of targeted drug delivery systems. Journal of Advanced Pharmaceutical Technology & Research 2010, 1, 374-80, 10.4103/0110-5558.76435.
- Vyas, J.; Puja, V.; Sawant, K. Formulation and Evaluation of Topical Niosomal Gel of Erythromycin. International Journal of Pharmacy and Pharmaceutical Sciences 2011, 3, 123–126.
- Vyas, J.; Vishal, G.; Tejas, G.; Vishal, C.; Umesh, U. Formulation and Characterization of Topical Gel of Erythromycin Entrapped into Niosomes. International Journal of PharmTech Research 2011, 3, 1714–1718.
- Mohammadi, S.; Farajzadeh, S.; Pardakhty, A.; Khalili Meybodi, M.; Mohebbi, A.; Yousefian, M.R.; Aflatoonian, M. A Survey to Compare the Efficacy of Niosomal Erythromycin Alone versus Combination of Erythromycin and Zinc Acetate in the Treatment of Acne Vulgaris. Journal of Kerman University of Medical Sciences 2017, 24, 420–430.
- Tao Liu; Ting Wu; Hongxi Liu; Bo Ke; Hongxing Huang; Zhenyou Jiang; Mingqiang Xie; Ultraviolet-crosslinked hydrogel sustained-release hydrophobic antibiotics with long-term antibacterial activity and limited cytotoxicity. Journal of Applied Polymer Science 2014, 131, 40438, 10.1002/app.40438.
- Rajib Basak; Ranjini Bandyopadhyay; Encapsulation of Hydrophobic Drugs in Pluronic F127 Micelles: Effects of Drug Hydrophobicity, Solution Temperature, and pH. Langmuir 2013, 29, 4350-4356, 10.1021/la304836e.
- Y Huang; Y Sun; Q Wang; Encapsulation and in vitro release of erythromycin using biopolymer micelle.. Cellular and molecular biology 2015, 61, 60-64.
- Huiling Song; Yu Yin; Jiahui Peng; Zixiu Du; Wei Bao; Preparation, Characteristics, and Controlled Release Efficiency of the Novel PCL-PEG/EM Rod Micelles. Journal of Nanomaterials 2021, 2021, 1-10, 10.1155/2021/8132868.
- Hanna Maria Gabriella Barriga; Margaret Nancy Holme; Molly M. Stevens; Cubosomes: The Next Generation of Smart Lipid Nanoparticles?. Angewandte Chemie International Edition 2019, 58, 2958-2978, 10.1002/anie.201804067.
- Sana Khan; Poorva Jain; Sourabh Jain; Richa Jain; Saurabh Bhargava; Aakanchha Jain; Topical Delivery of Erythromycin Through Cubosomes for Acne. Pharmaceutical Nanotechnology 2018, 6, 38-47, 10.2174/2211738506666180209100222.
- Séverine Jaspart; Géraldine Piel; Luc Delattre; Brigitte Evrard; Solid lipid microparticles: formulation, preparation, characterisation, drug release and applications. Expert Opinion on Drug Delivery 2005, 2, 75-87, 10.1517/17425247.2.1.75.
- Christian Joachim; To be nano or not to be nano?. Nature Materials 2005, 4, 107-109, 10.1038/nmat1319.
- Nicolae Strambeanu; Laurentiu Demetrovici; Dan Dragos; Mihai Lungu; Nanoparticles: Definition, Classification and General Physical Properties. Nanoparticles’ Promises and Risks: Characterization, Manipulation, and Potential Hazards to Humanity and the Environment 2014, null, 3-8, 10.1007/978-3-319-11728-7_1.
- Preshita P. Desai; Abhijit A. Date; Vandana B. Patravale; Overcoming poor oral bioavailability using nanoparticle formulations – opportunities and limitations. Drug Discovery Today: Technologies 2012, 9, e87-e95, 10.1016/j.ddtec.2011.12.001.
- Wolfgang Mehnert; Solid lipid nanoparticles Production, characterization and applications. Advanced Drug Delivery Reviews 2001, 47, 165-196, 10.1016/s0169-409x(01)00105-3.
- Sebastián Scioli Montoto; Giuliana Muraca; María Esperanza Ruiz; Solid Lipid Nanoparticles for Drug Delivery: Pharmacological and Biopharmaceutical Aspects. Frontiers in Molecular Biosciences 2020, 7, 587997, 10.3389/fmolb.2020.587997.
- Anil Kumar Sahu; Tekeshwar Kumar; Vishal Jain; Formulation Optimization of Erythromycin Solid Lipid Nanocarrier Using Response Surface Methodology. BioMed Research International 2014, 2014, 1-8, 10.1155/2014/689391.
- Pallavi Dhillon; Mohd. Aamir Mirza; Khalid Anwer; Abdullah Saud Alshetaili; Saad Maria Alshahrani; Zeenat Iqbal; Development and optimization of erythromycin-loaded lipid-based gel by Taguchi design: In vitro characterization and antimicrobial evaluation. Brazilian Journal of Pharmaceutical Sciences 2019, 55, e17395, 10.1590/s2175-97902019000217395.
- Pignatello, R.; Fuochi, V.; Petronio Petronio, G.; Greco, A.; Furneri, P.M. Formulation and Characterization of Erythromycin–Loaded Solid Lipid Nanoparticles. Biointerface Research in Applied Chemistry 2017, 7, 2145–2150
- Mumuni A. Momoh; Emmanuel C. Ossai; Omeje E. Chidozie; Omenigbo O. Precscila; Franklin C. Kenechukwu; Kenneth O. Ofokansi; Anthony A. Attama; Kunle O. Olobayo; A new lipid-based oral delivery system of erythromycin for prolong sustain release activity. Materials Science and Engineering: C 2018, 97, 245-253, 10.1016/j.msec.2018.12.041.
- Ifeanyi T. Nzekwe; Anselm C. Okere; Ifeanyi E. Okoye; Kokonne E. Ekere; Adaobi A. Ezenwa; Chukwuma O. Agubata; Bioassay-guided optimization of lipid-based erythromycin microparticles. Tropical Journal of Pharmaceutical Research 2020, 19, 1351-1358, 10.4314/tjpr.v19i7.2.
- Salvador Alvarez-Elcoro; Mark J. Enzler; The Macrolides: Erythromycin, Clarithromycin, and Azithromycin. Mayo Clinic Proceedings 1999, 74, 613-634, 10.4065/74.6.613.