Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Raziye Melike Yildirim.
Mitochondria are essential organelles and crucial for cellular survival. Mitochondrial biogenesis and mitophagy are dynamic features that are essential for both maintaining the health of the mitochondrial network and cellular demands. The accumulation of damaged mitochondria has been shown to be related to a wide range of pathologies ranging from neurological to musculoskeletal.
- mitophagy
- mitochondria
- Mitochondrial Homeostasis
1. Introduction
Mitochondria are essential organelles and are responsible for energy production for cellular survival. Therefore, dysfunctional or damaged mitochondria are related to a wide spectrum of pathologies, especially in tissues with the highest energy requirements [1]. Because mitochondria are crucial for the maintenance of the energy level of cells and for cellular survival, mitochondrial homeostasis is strictly regulated by mitochondrial biogenesis and mitophagy [2,3,4][2][3][4]. Mitophagy is the selective elimination process of damaged mitochondria by engulfment, and several molecular pathways are required for properly functioning mitophagy [5].
2. Mitochondrial Homeostasis
Mitochondria are essential organelles in cellular metabolism and physiology. Defective mitochondria are related to a broad spectrum of pathologies ranging from neurological to musculoskeletal [1]. Mitochondrial biogenesis and autophagy of mitochondria are two main pathways that preserve mitochondrial content and quality in cells. The tight regulation of these opposing pathways enables cells to adapt to the cellular energy demand and to respond to both intracellular and extracellular stress [12][6]. Mitochondria have their own circular genome that encodes 13 essential proteins for respiratory complexes. However, most mitochondrial proteins are encoded by nuclear DNA synthesized within the cytosol and imported into mitochondria. Several transcription factors are activated to control mitochondrial biogenesis in response to a variety of stimuli, including nutrition availability, hormones, growth factors, and temperature changes. Nuclear respiratory factors (NRF1 and NRF2), estrogen-related receptors (ERR-α, ERR-β, and ERR-γ), and the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) are among the key regulators of mitochondrial biogenesis. NRF1 and NRF2 drive the expression of the necessary nuclear genes encoding the proteins related to mitochondrial functioning. Additionally, transcription factor A (TFAM) and transcription factor B (TFB) proteins are important regulators of mitochondrial DNA transcription and replication and are regulated by both NRF1 and NRF2 [3,12,13][3][6][7] (Figure 1). Another group of nuclear hormone receptors including estrogen-related receptors ERR-α, ERR-β, and ERR-γ can stimulate mitochondrial biogenesis. They mainly regulate the transcription of genes involved in Krebs’ cycle, fatty acid oxidation, mitochondrial fusion, and fission. Furthermore, PGC-1α, a member of peroxisome proliferator-activated receptors (PPARs), serves as a coactivator and governs the activity of a wide range of transcription factors related to mitochondrial proliferation [4,12][4][6].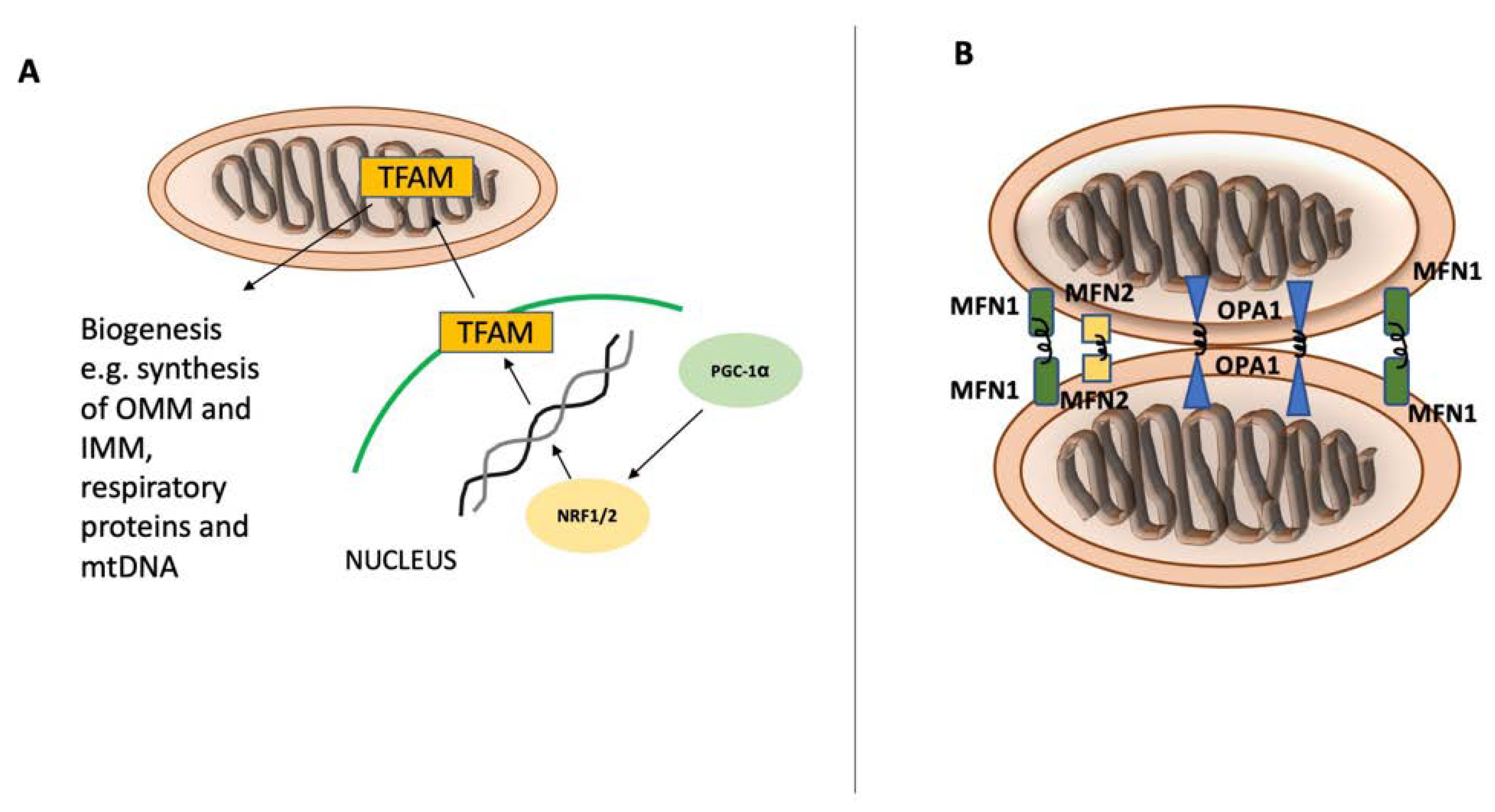
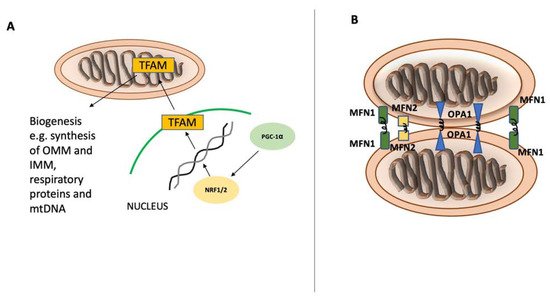
Figure 1. Mitochondrial biogenesis and fusion. Mitochondrial biogenesis and fusion. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is the main regulator of mitochondrial biogenesis (A) and translocates into the nucleus after its activation. In the nucleus, PGC-1α stimulates NRF1 and NRF2 expression, leading to increased activity of mitochondrial transcription factor A (TFAM), which orchestrates the synthesis of mtDNA and proteins. Mitochondrial fusion (B) is controlled by mitofusin-1 (MFN1) and mitofusin-2 (MFN2) on the mitochondrial outer membrane and by optic atrophy 1 (OPA1) on the inner mitochondrial membrane.
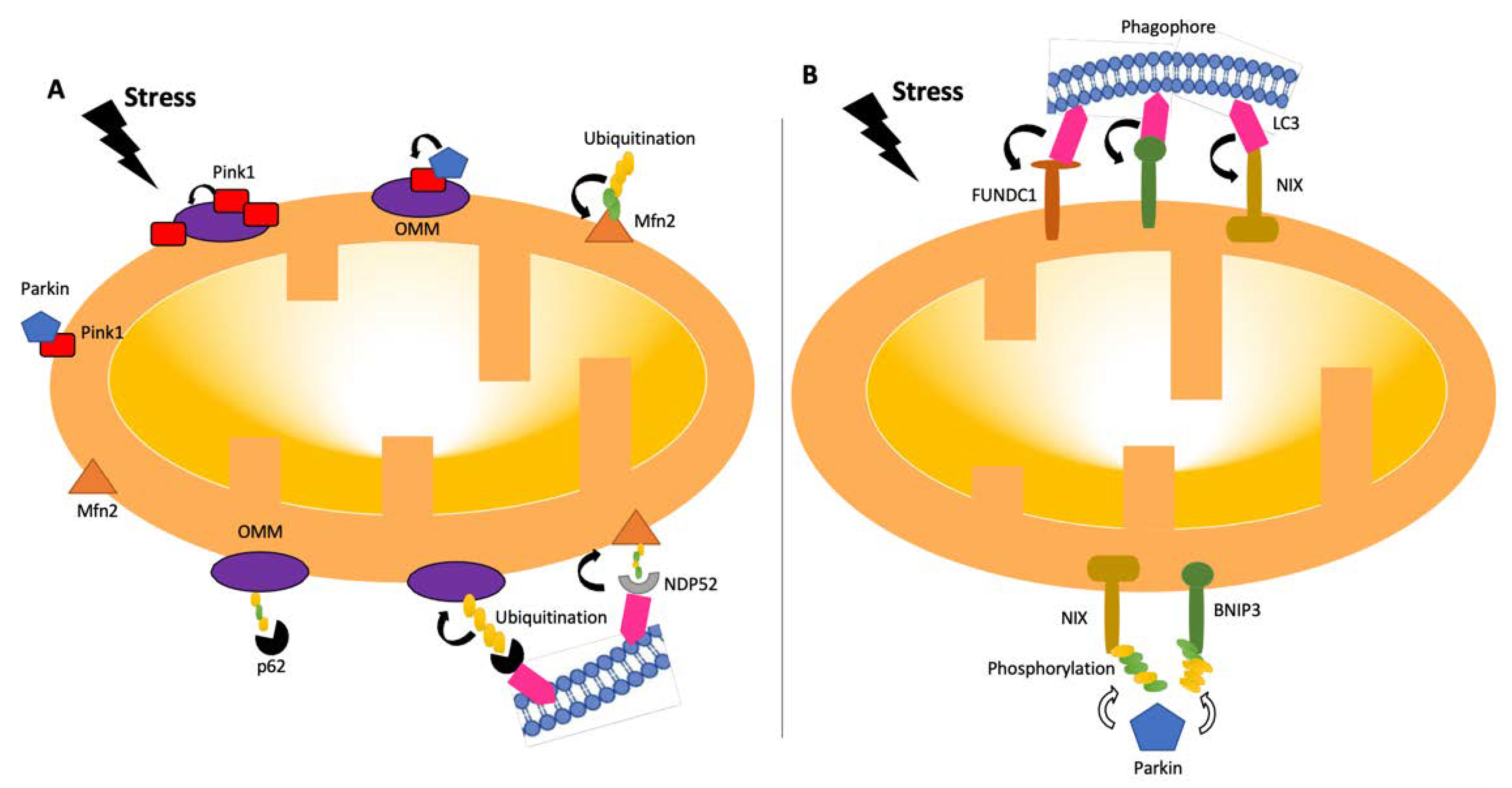
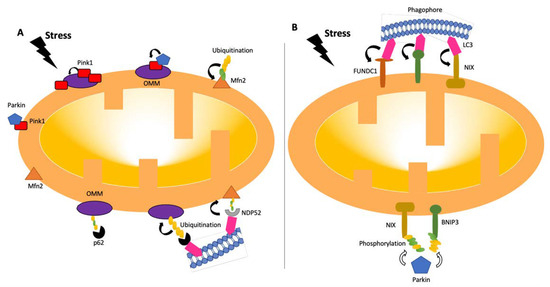
Figure 2. Pathways of mitophagy. Mitophagy pathways are either receptor- (B) or ubiquitin- (A) dependent [24,26][18][19]. Under stress conditions, mitochondria become dysfunctional, and PINK1 accumulates and stabilizes on the outer membrane of mitochondria, triggering PARKIN activation [27][20]. PARKIN recruits the ubiquitination and phosphorylation processes of outer-membrane proteins. Adaptor proteins, such as p62 and NDP52, recognize phosphorylated poly-ubiquitin chains. LC3 connects to adaptor proteins, and phagophores recognize LC3. In the receptor-mediated pathway (B), LC3 attaches to FUNDC1, BNIP3, or NIX receptors. BNIP3 or NIX can also be phosphorylated (PARKIN-mediated or -independent). LC3: microtubule-associated protein 1A/1B-light chain 3; BNIP3: Bcl-2/adenovirus E1B 19-kDa-interacting protein 3; FUNDC1: mitochondrial receptor FUN14 domain-containing 1; NIX: BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like.
References
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174.
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022.
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638.
- Dominy, J.E.; Puigserver, P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a015008.
- Rodger, C.E.; McWilliams, T.G.; Ganley, I.G. Mammalian mitophagy—From in vitro molecules to in vivo models. FEBS J. 2018, 285, 1185–1202.
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188.
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366.
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42.
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546.
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951.
- Kirkin, V.; Rogov, V.V. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol. Cell 2019, 76, 268–285.
- Johansen, T.; Lamark, T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J. Mol. Biol. 2020, 432, 80–103.
- Gubas, A.; Dikic, I. A guide to the regulation of selective autophagy receptors. FEBS J. 2022, 289, 75–89.
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 2016, 129, 2170–2181.
- Pryde, K.R.; Smith, H.L.; Chau, K.-Y.; Schapira, A.H. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 2016, 213, 163–171.
- Wu, W.; Lin, C.; Wu, K.; Jiang, L.; Wang, X.; Li, W.; Zhuang, H.; Zhang, X.; Chen, H.; Li, S. FUNDC 1 regulates mitochondrial dynamics at the ER–mitochondrial contact site under hypoxic conditions. EMBO J. 2016, 35, 1368–1384.
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185.
- Fritsch, L.E.; Moore, M.E.; Sarraf, S.A.; Pickrell, A.M. Ubiquitin and Receptor-Dependent Mitophagy Pathways and Their Implication in Neurodegeneration. J. Mol. Biol. 2020, 432, 2510–2524.
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080.
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002.
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91.
- Sekine, S. PINK1 import regulation at a crossroad of mitochondrial fate: The molecular mechanisms of PINK1 import. J. Biochem. 2020, 167, 217–224.
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108.
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166.
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–141.
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375.
- Villa, E.; Proics, E.; Rubio-Patino, C.; Obba, S.; Zunino, B.; Bossowski, J.P.; Rozier, R.M.; Chiche, J.; Mondragon, L.; Riley, J.S.; et al. Parkin-Independent Mitophagy Controls Chemotherapeutic Response in Cancer Cells. Cell Rep. 2017, 20, 2846–2859.
- Orvedahl, A.; Sumpter, R., Jr.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117.
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242.
- Kubli, D.A.; Quinsay, M.N.; Huang, C.; Lee, Y.; Gustafsson, A.B. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2025–H2031.
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890.
More