Substance use disorders (SUDs) are characterized by compulsive drug use despite clinically significant distress and other negative consequences in life. Substance use disorders alter how the brain and body respond to addictive substances, and patients with SUDs suffer from an inability to quit addictive substances. Addictive substances comprise natural, semi-synthetic, and synthetic substances, such as amphetamine/methamphetamine, cocaine, opioids, cannabinoids, alcohol, hypnotics/anxiolytics, inhalants, nicotine, and caffeine. Addictive-substance misuse and problems that are associated with SUDs have been a serious societal concern worldwide.
2. Fundamental Function of GIRK Channels and Response to Addictive Substances
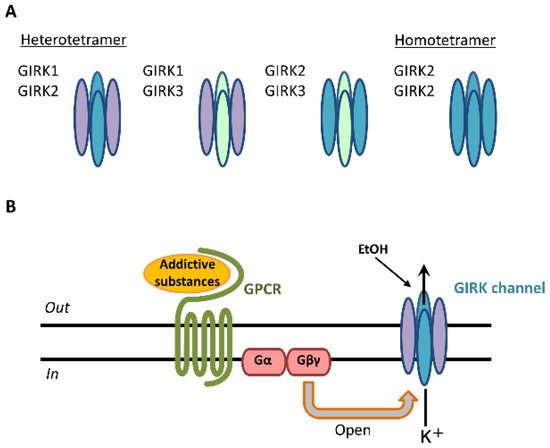
GIRK channels are encoded by the
Kcnj gene and assembled in homotetrameric or heterotetrameric units comprising four subunits: GIRK1 (Kir3.1/Kcnj3), GIRK2 (Kir3.2/Kcnj6), GIRK3 (Kir3.3/Kcnj9), and GIRK4 (Kir3.4/Kcnj5)
[1][2][3][2,3,4]. Each GIRK subunit possesses two transmembrane domains, TM1 and TM2, that flank a hydrophobic pore domain and intracellular N- and C-terminal domains
[3][4]. GIRK1-3-containing channels are widely expressed in the rodent brain, including the cerebral cortex, amygdala, hippocampus, thalamus, ventral tegmental area (VTA), locus coeruleus, and cerebellum. GIRK channels are expressed in the brain’s reward system
[4][5][5,6]. The expression of GIRK4 subunits is found mainly in the heart and limited to only a few brain regions, such as the neocortex, insular cortex, cerebellar cortex, hypothalamus, thalamus, and brainstem
[6][7][7,8]. Three primary GIRK channel subunits in the brain form heteromeric channels: GIRK1/GIRK2, GIRK1/GIRK3, and GIRK2/GIRK3
[3][4] (
Figure 1A).
Figure 1. GIRK channel subunits in the brain and signal transduction of addictive substances through GIRK channels. Three types of GIRK subunits are expressed and form heterotetramers (GIRK1/GIRK2, GIRK1/GIRK3, GIRK2/GIRK3) and homotetramers (GIRK2/GIRK2) in various brain regions (A). Addictive substances or neurotransmitters bind to GPCRs, and the GIRK channel is activated by the G protein Gβγ subunit. Following this activation, potassium ions (K+) flow into the cell, and cellular excitability decreases (B).
GIRK2 subunits can uniquely form functional homomers in the brain
[3][4]. These various compositions exhibit K
+ selectivity, inward rectification, and G-protein-dependent gating
[1][3][2,4]. Outward K
+ currents through GIRK channels inhibit cellular excitability
[1][3][2,4]. The core reward circuitry in the brain consists of the VTA, nucleus accumbens (NAc), and ventral pallidum via the medial forebrain bundle. The VTA is the initiating nucleus of the dopaminergic system, which then projects to the NAc via the medial forebrain bundle
[8][9]. Dopamine neurons in the VTA also project to the amygdala, orbitofrontal cortex, anterior cingulate cortex, hippocampus, and prefrontal cortex
[9][10]. Dopamine neurons in the VTA express only the GIRK2 and GIRK3 subunits, whereas γ-aminobutyric acid (GABA) neurons in the VTA express the GIRK1, GIRK2, and GIRK3 subunits
[4][5][5,6]. Addictive substances enhance reward circuitry in the brain and produce feelings of pleasure. These “rewarding effects” positively reinforce their use and increase relapse risk. Addictive substances include alcohol, nicotine, caffeine, psychostimulants (amphetamines, methamphetamines), cocaine, and opioids
[10][11]. Activation by these substances is mediated by interactions with G-protein-coupled receptors (GPCRs), including GABA receptors,
N-methyl-D-aspartate (NMDA) receptors, nicotinic acetylcholine receptors, adenosine receptors, and opioid receptors, as well as dopamine transporters
[9][11][12][13][14][15][16][17][18][10,12,13,14,15,16,17,18,19]. Following the stimulation of GPCRs, GIRK channels are activated through coupling of the G
i/o family of G-proteins to those receptors and gated by G-protein βγ subunits that are released from the G-protein α subunit
[19][20][20,21] (
Figure 1B). G-protein βγ subunits activate GIRK channels through direct binding with amino- and carboxyl-ends of the channels
[1][21][2,22]. The G-protein α subunit was not thought to be responsible for GIRK channel activation, but rather to be a regulator of the specificity of channel activation
[3][22][4,23].
GIRK1 knockout and GIRK2 knockout mice exhibited baseline hyperactivity and an increase in locomotor response to cocaine
[23][27]. GIRK1 knockout and GIRK2 knockout mice exhibited a decrease in analgesic responses after morphine administration into the spinal cord
[24][28]. GIRK2 knockout and GIRK3 knockout mice also exhibited a decrease in cocaine self-administration
[25][30]. GIRK2 knockout mice did not exhibit alcohol-induced conditioned taste aversion or conditioned place preference
[26][31]. Morphine-induced activity increased in GIRK1 knockout and GIRK2 knockout mice but decreased in GIRK3 knockout mice compared with wildtype mice
[27][29]. GIRK3 knockout mice exhibited a reduction of alcohol withdrawal
[6][7] and an alcohol-induced conditioned place preference
[28][37]. Additionally, GIRK3 knockout mice exhibited a selective increase in alcohol binge-like drinking without affecting alcohol metabolism or the sensitivity to alcohol intoxication
[29][38]. The roles of GIRK channels have also been studied using
weaver mutant mice, which have spontaneously occurring autosomal recessive mutations of the
Girk2 gene that lead to a reduction of GIRK2 channel function and cause abnormalities of dopamine signaling
[30][39].
Weaver mutant mice exhibited lower antinociceptive effects of alcohol
[31][24] and opioids
[32]. Amphetamine caused less hyperlocomotion in
weaver mutant mice
[33].
Weaver mutant mice also did not exhibit methamphetamine-induced conditioned place preference or priming effects
[34]. A significant decrease in basal and methamphetamine-induced dopamine release was also detected in the NAc, with a decrease in methamphetamine-induced neural activity in the posterior NAc shell
[34]. Furthermore, neuron-specific knockout mice have been generated to investigate the role of GIRK channels in the reward system. McCall et al. (2017) reported that the genetic ablation of GIRK2 in dopamine neurons, which did not alter the baseline excitability of VTA dopamine neurons, increased behavioral sensitivity to cocaine
[35]. The overexpression of GIRK3 in VTA dopamine neurons decreased GABA
B receptor- and dopamine D
2 receptor-dependent signaling and increased cocaine-induced locomotion, whereas the overexpression of GIRK2 increased GABA
B receptor-dependent signaling and decreased cocaine-induced locomotion
[36]. Knockout and missense mutation mice are useful for studying the response of GIRK channels to addictive substances, but the response to addictive substances may differ between mice with knockouts and missense mutations. This may be attributable to the complete or functional deficiency of GIRK channels. The relevance of GIRK channel function in VTA dopamine neurons has been demonstrated, and GIRK channels have been shown to play a key role in behavioral responses to addictive substances.
3. Pharmacological Modulation of GIRK Channels and Therapeutic Effects
Although GIRK channels are activated in a G-protein-dependent or -independent manner by addictive substances, antipsychotic compounds, psychoactive compounds (e.g., antidepressants, including selective serotonin re-uptake inhibitors), and a cerebral circulation/metabolism ameliorator were shown to block brain-type GIRK1/GIRK2 channels and cardiac-type GIRK1/GIRK4 channels in the
Xenopus oocyte expression assay
[37][38][39][40][41][42][42,43,44,45,46,47]. Interestingly, fluoxetine, desipramine, paroxetine, sertraline, and ifenprodil blocked alcohol-induced GIRK1/GIRK2 currents
[38][39][40][41][42][43,44,45,46,47]. Ifenprodil can also inhibit GIRK channels at a lower IC
50 than antidepressants (e.g., fluoxetine, imipramine, desipramine, amitriptyline, nortriptyline, clomipramine, maprotiline, paroxetine, sertraline, duloxetine, and amoxapine)
[38][39][40][41][42][43,44,45,46,47]. Each GIRK channel was inhibited by ifenprodil at the following IC
50 values: 7.01 ± 0.92 μM at GIRK1/GIRK2, 8.76 ± 1.26 μM at GIRK2, and 2.83 ± 0.69 μM at GIRK1/GIRK4
[41][46]. Although these compounds do not have specificity for GIRK channels, some of these compounds were shown to inhibit behaviors that are induced by addictive substances. Takamatsu et al. reported that pretreatment with fluoxetine and paroxetine inhibited methamphetamine-induced conditioned place preference in mice, which was unaffected by fluvoxamine, an antidepressant that does not block GIRK channels
[43][44][48,49]. However, paroxetine has several adverse effects, including serotonin syndrome, neuroleptic malignant syndrome, convulsions, toxic epidermal necrosis, antidiuretic hormone incompatible secretion syndrome, severe liver dysfunction, rhabdomyolysis, low white blood cell counts, and anaphylaxis
[45][50]. The U.S. Food and Drug Administration (FDA) approved fluoxetine, but it is not currently approved for use in Japan. Ifenprodil is a blocker of α
1-adrenergic receptors, GluN2B subunit-containing NMDA receptors
[46][47][51,52], and sigma-1/2 receptors
[48][53], and also inhibits GIRK channels
[41][46][46,51]. Ifenprodil inhibited methamphetamine-induced conditioned place preference
[49][54], and pretreatment with ifenprodil reduced morphine-induced conditioned place preference in mice
[50][55]. Ifenprodil also inhibited the amphetamine-induced potentiation of excitatory postsynaptic currents in rat midbrain dopamine neurons
[51][56]. Pretreatment with a combination of ifenprodil and cyproheptadine in mice did not cause locomotor sensitization compared with mice that were pretreated with saline when the mice were repeatedly injected with D-amphetamine
[52][57]. Notably, ifenprodil inhibits GluN2B subunit-containing NMDA receptors at a lower IC
50 than GIRK channels (IC
50: 0.34 μM at GluN1A/GluN2B receptors; see IC
50 values for GIRK channels above)
[46][51]. A recent study reported that the methamphetamine-induced increase in locomotor activity (i.e., behavioral sensitization) was blocked by ifenprodil via GluN2B-protein phosphatase 2A-AKT signaling in the dorsal striatum in mice
[53][58]. These studies suggest that ifenprodil inhibits behaviors that are induced by addictive substances through GluN2B subunit-containing NMDA receptors. Thus, ifenprodil may exert effects on behaviors that are caused by addictive substances through NMDA receptors and/or GIRK channels. Kotechi et al. reported that GIRK2/GIRK3 channels in VTA dopamine neurons regulated morphine-induced motor activity, whereas GIRK channel activation in VTA GABA neurons was not required
[27][29]. GIRK channels play a key role in the influence of addictive substances on the reward system, but further studies are warranted to define the mechanisms of action of ifenprodil.