Unlike animals, plants are immobile and could not actively escape the effects of aggressive environmental factors, such as pathogenic microorganisms, insect pests, parasitic plants, extreme temperatures, drought, and many others. To counteract these unfavorable encounters, plants have evolved very high phenotypic plasticity. In a rapidly changing environment, adaptive phenotypic changes often occur in time frames that are too short for the natural selection of adaptive mutations. Probably, some kind of epigenetic variability underlines environmental adaptation in these cases. Indeed, isogenic plants often have quite variable phenotypes in different habitats. There are examples of successful “invasions” of relatively small and genetically homogenous plant populations into entirely new habitats. The unique capability of quick environmental adaptation appears to be due to a high tendency to transmit epigenetic changes between plant generations. Multiple studies show that epigenetic memory serves as a mechanism of plant adaptation to a rapidly changing environment and, in particular, to aggressive biotic and abiotic stresses. In wild nature, this mechanism underlies, to a very significant extent, plant capability to live in different habitats and endure drastic environmental changes. In agriculture, a deep understanding of this mechanism could serve to elaborate more effective and safe approaches to plant protection.
- plant epigenetics
- epigenetic variability
- abiotic stress
- biotic stress
- environmental adaptation
- gene expression
- DNA methylation
- chromatin
- siRNA
1. Introduction
- Introduction
Plants live in a constantly changing environment that is often unfavorable or even hostile. As sessile organisms, plants cannot actively escape multiple aggressive encounters. Instead, they developed high phenotypic plasticity that includes rapid responses to aggressive environmental factors and adaptations to changing environments. Changes in gene expression underlie this phenotypic plasticity. Since epigenetic marks control gene expression, the epigenetic variation could be a key player in plant responses to stress factors and environmental adaptation.
The most thoroughly studied type of epigenetic phenomena in plants is DNA methylation [1][2][1,2]. A major part of methylated cytosine residues (m5C) in plants, like in animals, occurs in the symmetric CG sites. Unlike animals, plants also display significant methylation in the symmetric CHG sites and asymmetric CHH sites (H is any nucleotide except G). All three methylation contexts are present in repeat and transposable element (TE) sequences, while the protein-coding gene sequences are mostly methylated at CG sites. The maintenance methylation of CG sites is carried out by DNA methyltransferase MET1 with the assistance of three VIM (VARIANT IN METHYLATION) family proteins, VIM1–VIM3 [3]. This methylation complex recognizes and methylates with a high preference hemi-methylated CG sites in the daughter strands during DNA replication. A plant-specific DNA methyltransferase CMT3 (CHROMOMETHYLASE 3) is responsible for the maintenance methylation of symmetric CHG sites. Unlike MET1, CMT3 cannot recognize hemi-methylated sites and maintains CHG-specific methylation due to mutual stimulating substrate-level interactions between CMT3 and H3K9-specific histone methyltransferases (HMTs) [4]. DNA methylation de novo at CG, CHG, and CHH sites occurs mainly by the DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2) [5]. Due to the asymmetric nature of CHH sites, their methylation is maintained by recurring methylation de novo. Recently, an alternative pathway dependent on CMT2 was found to participate in CHH methylation [6][7][6,7]. Similar to CMT3, CMT2 is targeted to methylated sites via histone H3K9 methylation marks. In contrast, DRM2 is targeted to its methylation sites due to the complementary interaction of 24-nt siRNAs (24-nucleotide small interfering RNAs) with sequences to be de novo methylated – the RNA-directed DNA methylation (RdDM) pathway [8][9][8,9].
In Arabidopsis, DNA could be actively demethylated via a base excision repair pathway involving the activity of dedicated m5C-specific glycosylase enzymes REPRESSOR OF SILENCING 1 (ROS1), DEMETER (DME), DEMETER-LIKE 2 (DML2), and DEMETER-LIKE 3 (DML3) [10]. It has been shown that DNA methylation status in multiple genome loci is the net result of their recurrent methylations by RdDM and demethylations by ROS1 [11].
Besides DNA methylation, plants and other eukaryotic organisms have another set of epigenetic marks – covalent modifications of histone proteins' various amino acid residues. Among the plethora of histone modifications, several types of methylation at lysine residues were best studied both in animals and plants [12][1,12]. In Arabidopsis, three types of H3K4 methylation marks (mono/di/tri-methylation – H3K4me1, H3K4me2, and H3K4me3) are found at gene bodies (H3K4me1) and promoters (H3K4me2 and H3K4me3) of actively transcribed genes [13]. H3K4me2/3 and m5C are mutually exclusive marks at the same promoter, while H3K4me1 could coexist with m5C along the gene bodies.
H3K27me3 shows a robust correlation with the repression of gene transcription at specific loci. Multiple genes are known to be regulated by this epigenetic mark during plant development, mostly independent of other epigenetic mechanisms [14]. Regulation of gene transcription by H3K27me3 marks is mediated by the Polycomb Repressive Complex 2 (PRC2) that includes an H3K27-specific histone methyltransferase (HMT). The plant chromodomain protein LIKE HETEROCHROMATIN PROTEIN 1 (LHP1) binds to H3K27me3-containing genome loci and probably participates in mediating its regulatory effects [15].
H3K9me2 is another robustly repressive mark in plants. Unlike H3K37me3, this epigenetic mark is essentially heterochromatin-specific. As critical partners in non-CG DNA methylation by CMT3 and CMT2, H3K9me2 marks mostly colocalize with methylated CHG and CHH sites in repeat- and transposon-rich genome compartments [16].
Different kinds of small RNAs (sRNAs) in plants act via recognition of complementary sequences in mRNA or DNA, leading to posttranscriptional gene silencing (PTGS) due to the degradation of targeted mRNAs or inhibition of their translation (miRNAs and 21-22-nt siRNAs) or transcriptional gene silencing (TGS) due to DNA methylation via RdDM pathway (24-nt siRNAs) [17].
2. Epigenetic responses to stressful factors
- Epigenetic responses to stressful factors
2.1. Abiotic stress
Abiotic stresses mainly include extreme cold, heat shock, water deficit, excessive salinity, nutrient deficiencies, and heavy metal toxicity. To study pure (not caused by genetic factors) epigenetic variability, genetically uniform populations are usually used. When identical cloned lines of apomictic dandelion plants were exposed to various stresses, individual plants in all groups displayed significant variations in DNA methylation [18]. Smaller variations were observed in control (unexposed) group. These variations were mostly heritable, and new variations often arose in daughter plants. These data show that environmental stress increases epigenetic variability mostly through stochastic epigenetic changes.
2.1.1. Cold stress
Cold stress has profound effects on plant metabolism and gene expression. When exposed to low non-freezing temperatures, plants display increased tolerance to subsequent freezing temperatures – a phenomenon known as cold acclimation. Cold stress increases the levels of C-repeat binding factor family proteins (CBFs) – transcription factors that upregulate multiple cold-responsive (COR) effector genes [19]. PICKLE (PKL) is a subunit of the Mi-2/CHD3 subfamily of ATP-dependent chromatin remodelers that affects cold acclimation through the modulation of the CBF3 functional activity [20]. More than 600 genes were differentially expressed between the wild-type and pkl mutant plants after cold treatment, including the downregulation of CBF3 and multiple CBF target genes, such as RD29A, COR15A, and COR15B. Since PKL is known to be involved in the RdDM [21] and H3K27me3 deposition [22] pathways, both H3K27me3 and DNA methylation could serve as memory marks for cold-induced freeze tolerance.
In Arabidopsis, WD40 repeat-containing protein HOS15 functions as a targeting protein in the ubiquitination-proteasome degradation pathway, while HISTONE DEACETYLASE 2C (HD2C) is one of its interacting partners [23]. The histone H3 deacetylating activity of HD2C negatively regulates the expression of genes involved in cold acclimation, while HOS15 counteracts this negative regulation by mediated proteasomal degradation of HD2C at COR gene promoters.
2.1.2. Heat stress
In heat shock (HS) response, heat shock transcription factors A1 (HsfA1s) serve as “master regulators” that activate multiple transcriptional networks [24]. HsfA1s directly regulate the transcription of genes coding for essential HS-responsive transcription factors (TFs). Unlike animals, plants evolved extensive families of HS factors (HSFs) that differ in their expression patterns and functions. As master regulators, HsfA1s are indispensable in the HS response.
A study using a set of epigenetic mutants showed that the RdDM pathway and the Rpd3-type histone deacetylase HDA6 play important and independent roles in basal heat tolerance [25]. HS induces the sustained accumulation of H3K9Ac and H3K4me3 on various heat shock protein genes [26].
2.1.3. Salt stress
By Na+ ion toxicity, hyperosmotic stress, and oxidative damage, high salinity greatly impacts plant growth and development. Evaluation of global DNA methylation levels in rice varieties largely different in salt tolerance found reduced DNA methylation in leaves after exposure to salt stress [27]. In the salt-tolerant variety, the reduction in global DNA methylation is rapid and profound. In contrast, in the salt-susceptible variety, the methylation loss is small and not statistically significant.
In roots, the salt stress-induced DNA methylation changes are mostly demethylation. A substantial share of these DNA methylation changes is stable throughout the recovery period when the stress is removed [28]. Since the genome sequences of the salt-tolerant and salt-sensitive cultivars in this study are very similar, stable DNA methylation differences (epialleles) may be responsible for phenotypic variations, including different salinity tolerance.
In plants, Ca2+‐CALCINEURIN B‐LIKE PROTEIN (CBL)‐CBL INTERACTING PROTEIN KINASE (CIPK) complex participates in the regulation of cellular ion homeostasis [19]. High Na+, low K+, excess Mg2+, and high pH cause cytosolic Ca2+ signals, which activate the SOS pathway, including SOS1 (Na+/H+ antiporter), AKT1 (K+ channel), Mg2+ transporter, and H+ ATPase. HIGH‐AFFINITY K+ CHANNEL 1 (HKT1) mediates Na+ influx and, together with the SOS pathway, determines salinity tolerance in plants. In Arabidopsis, a putative siRNA target region at ~2.6 kb upstream of the HKT1 gene start codon is heavily methylated in all sequence contexts in the wild-type plants [29]. The DNA methylation-deficient mutant met1 is hypersensitive to salt stress. Heavy methylation of the HKT1 promoter inhibits transcription in leaves and roots. In contrast, non-CG methylation fine-tunes the expression of HKT1 in leaves, which may be essential in the long-term adaptation of plants to salt stress, but not in the short term salt tolerance. This DNA methylation-dependent regulation mechanism could be essential to balance HKT1 expression between leaves and roots. Besides DNA methylation, histone acetylation regulates plant adaptation to high-salinity stress [30].
After a recovery period, Arabidopsis plants primed by exposure to mild salt stress display less salt uptake and higher drought tolerance than control plants [31]. Specific changes in the H3K27me3 profiles occur under the salt treatment and are maintained over a 10-day recovery period. The vast majority of differentially methylated H3K4me2 and H3K4me3 sites have higher methylation levels in the primed plants. By contrast, the vast majority of differential H3K27me3 sites have lower methylation levels in the primed plants. These data indicate a more open chromatin configuration in primed plants without major changes in genome-wide histone modification profiles. The lower shoot salt accumulation in primed plants upon the second salt treatment mimics the phenotype of mutant plants over-expressing HKT1. In primed plants, increased HKT1 mRNA levels are consistently observed after the second salt treatment at 10 d. Thus, HKT1 is a prime candidate for explaining at least one of the priming physiological effects.
R2R3-MYB is the largest subfamily of the MYB family TFs known to regulate plants' defense responses [32][34][32]. In Arabidopsis, MYB74 directly regulates the expression of the salinity stress genes. Significant DNA methylation in CG and CHH contexts and siRNA target sites are present in the MYB74 promoter region, while a noticeable reduction in methylation occurs upon the salt treatment. The MYB74 mRNA level increases about eightfold under salt stress, in a close correlation with promoter demethylation, and the accumulation of 24-nt siRNAs that target MYB74 promoter substantially reduces. The decrease in DNA methylation and induction of MYB74 transcription under salt stress is probably due to the reduction of these 24-nt siRNAs.
2.1.4. Water deficit stress
Most plants encounter water deficit stress many times across their lifespan. Multiple mechanisms help plants withstand these recurring drought encounters, including stress memory [33][33][33,34]. Abscisic acid (ABA) plays a vital role in regulating the activity of multiple drought stress-responsive genes. In Arabidopsis, repeated dehydration upregulates several ABA-induced genes [35][36][37][38][35–38]. Moreover, the guard cell-specific memory maintains partially closed stomata across the recovery period [38].
Water deficit increases ABA production, which promotes the increased resistance to water deficit [39]. The H3K4me3-specific methylase ATX1 stimulates the transcription of multiple genes involved in responses to biotic and abiotic stresses, including drought stress [35]. The drought stress tolerance is accordingly diminished in the atx1 mutant compared with the wild-type plants. This higher sensitivity of the atx1 mutant plants to the water deficit is explained by their leaves' more rapid transpiration due to higher stomatal apertures. The ABA levels in atx1 plants are only 40% of those in the wild-type plants. Of the four ABA biosynthetic genes, NCED3, supposedly the rate-limiting factor in ABA biosynthesis, shows diminished expression under dehydration stress in atx1 compared with wild-type plants. ATX1 binds to a promoter region of the NCED3 gene, and the levels of H3K4me3 at the NCED3 promoter region are increased by dehydration stress, much higher in the wild-type than the atx1 mutant plants. The H3K4me3 levels at multiple dehydration stress-responsive genes show a good correlation with their transcript levels, and genes downregulated in atx1 plants show reduced levels of H3K4me3.
The transcriptional responsiveness of genes induced by water deficit correlates with changed histone modifications and nucleosome density [30]. Intense dehydration stress leads to a more pronounced increase in H3K4me3 and H3K9ac and a decrease in nucleosome density on inducible genes compared with moderate dehydration. Thus, epigenetic responsiveness appears to depend on the intensity of the stress. During recovery from stress, H3K9ac rapidly decreases, and RNA polymerase II is removed from the drought stress-upregulated genes, while H3K4me3 decreases gradually upon rehydration.
In Arabidopsis, LHP1 is a component of the repressive complex PRC1 that binds to H3K27me3 marks via its chromodomain. The binding of LHP1 to ABA-responsive genes ANAC019, ANAC055, and VSP1 and their H3K27me3 levels decreases after ABA treatment [40]. Thus, LHP1 contributes to their repression via increased H3K27me3 marks. ANAC019 and ANAC055 are known as positive regulator TFs of drought tolerance. The lhp1 mutant plants show increased ABA sensitivity and significantly higher tolerance to a prolonged drought than wild-type plants. Therefore, LHP1 negatively regulates ABA-mediated responses to drought, probably via increased H3K27me3 at ANAC019 and ANAC055.
The cumulative effect of multigenerational drought stress on genome-wide DNA methylation was studied in drought-sensitive and drought-resistant rice cultivars [41]. A larger proportion of differentially methylated loci showed transgenerational inheritance in the drought-tolerant rice cultivar. These findings could have important implications in understanding the place of epigenetic variation in plant evolution.
2.1.5. Nutrient deficits stress
Very much like any other environmental factor, the nutrients are perceived by multiple signaling pathways assisting in plant adaptation to their fluctuating availability in the soil [42]. High nitrogen (N) represses the expression of a root nitrogen transporter, NRT2.1, via a negative feedback loop mediated by HNI9 (High Nitrogen Insensitive 9) – a critical factor in the deposition of repressive H3K27me3 marks at the NRT2.1 gene [43].
The iron homeostasis is negatively regulated by the PRMT5‐mediated symmetric dimethylation of the histone H4 third arginine residue (H4R3sme2) at several genes of the bHLH family subgroup Ib [44]. In the PRMT5 deficient mutant plants, higher iron accumulation in shoots and greater iron deficiency tolerance are observed relative to the wild-type plants. In Arabidopsis, a mutation in GCN5 (General Control Nonrepressed protein5) was found to impair the iron translocation from the root to the shoot [45]. The expression of GCN5 reaches a maximal level at 3 d of iron-deficiency treatment, being upregulated by more than fivefold over the control. Five genes related to iron transport, FRD3, EXO70H2, MLP329, BOR1, and CRK25, are direct targets of GCN5. Consistent with the known function of GCN5 as a histone acetyltransferase, the H3K9ac and H3K14ac and mRNA levels of these five genes under iron-deficiency conditions are significantly lower in gcn5 mutant than wild-type plants.
The AL6 (Alfin Like 6) gene plays an essential role in root hair formation induced by phosphate starvation and several other processes related to cellular phosphate homeostasis [46]. Since AL6 is known to be a PHD finger reader protein of H3K4me3 epigenetic marks, these data indicate a possible role of H3K4me3 and other chromatin marks in plant adaptation to phosphate deficit. The phosphate (Pi) starvation induces multiple changes in DNA methylation in Arabidopsis and rice [47][48][47,48].
2.2. Biotic stress
2.2.1.
Viruses
Among the first evidence for epigenetic regulation of plant tolerance towards biotic factors was the control of viral virulence via PTGS [40]. Upon infection by RNA viruses, plants recognize viral double-strand RNA molecules, inducing their degradation into siRNAs. Another mechanism, TGS, provides for a more permanent defense against DNA viruses via RdDM. Unlike animals, plants do not have adaptive immune systems. Instead, they evolved a most complex RNA-based system of gene regulation and protection against foreign nucleic acids. Most plant viruses have a single-stranded RNA (ssRNA) genome. It was recently shown that m6A-specific methylation of the RNA genome in the alfalfa mosaic virus (AMV) controls Arabidopsis infection [49]. The Arabidopsis protein ALKBH9B is an m6A demethylase involved in mRNA silencing and/or mRNA decay processes. ALKBH9B was shown to affect the infectivity of AMV but not of cucumber mosaic virus (CMV), correlating with the ability of ALKBH9B to bind or not to their coat proteins.
2.2.2. Microbes
The role of DNA methylation in plant immunity has been exhaustively studied [50][40,50]. The first layer of active defense, known as pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI), relies on the perception of PAMPs or microbe-associated molecular patterns (MAMPs) by pattern-recognition receptors (PRRs). PAMP perception is followed by the activation of immune responses, which results in basal immunity. To override the plant defense, pathogens produce special effector molecules that damp PTI. As a counter-counter defense, these pathogen effectors could be perceived by disease resistance proteins, often resulting in a potent immune response – effector-triggered immunity (ETI). Activation of both PTI and ETI involves massive changes in gene expression regulated by epigenetic mechanisms.
Flagellin Sensing 2 (FLS2) is a well-characterized plant PRR that senses the bacterial flagellin-derived peptide flg22, leading to changed expression of multiple genes [51]. The flg22 treatment was shown to reactivate TEs and other well-characterized RdDM targets, suggesting that it inhibits TGS. Significant downregulation of the key components of the RdDM pathway occurs at 3 h and 6 h after flg22 treatment, which correlates with the upregulation of the early defense gene Flg22-induced Receptor-Kinase 1 (FRK1). The majority of the RdDM components return to normal levels at 9 h concomitant with induction of the late defense gene Pathogenesis-related gene 1 (PR1). Moderately increased bacterial growth in ros1 mutant plants infected with Pst strain DC3000 supports the role of active DNA demethylation in antibacterial resistance.
Plant NOD-like receptors (NLRs) are key immune receptors whose overexpression triggers a constitutive hypersensitive response (HR) and/or PR1 induction. The HR-like phenotype and enhanced PR1 expression are observed in met1 nrpd2 double mutant plants, suggesting that some NLRs might be directly controlled by RdDM.
The innate immunity responses in plants are often short-term but can elicit the acquired immunity state that manifests itself as “priming” of inducible defenses [52]. Primed plants respond faster and/or stronger to recurring defense stimuli. Priming could be induced by microbes, as in pathogen-induced systemic acquired resistance (SAR). Other priming states can be triggered by chemicals, such as β-aminobutyric acid (BABA). Some priming states are relatively short-term and disappear within a few days, while others are long-lasting and can even be transmitted between plant generations. The priming of salicylic acid (SA)-dependent immunity is long-lasting and transgenerationally inheritable, suggesting the involvement of epigenetic mechanisms.
In potato, priming with BABA increases resistance to the oomycete pathogen Phytophthora infestans, the causal agent of late blight disease [53]. The first unstressed generation of the BABA-primed parent plants shows increased resistance to the P. infestans, probably due to the upregulation of SA-responsive genes. During the early priming phase, a bivalent histone mark configuration, H3K4me2 and H3K27me3, is observed on the SAR regulator genes NPR1 (Non-expressor of PR genes) and SNI1 (Suppressor of NPR1, Inducible). This bivalent chromatin configuration is readily switchable between active and silent states, and the increased responsiveness of the PR1 and PR2 genes contributes to the intergenerational stress memory.
2.2.3. Pests
The soybean cyst nematode (SCN; Heterodera glycines) penetrates soybean roots to induce the formation of a multinucleated feeding site – the syncytium. Genome hypomethylation in all sequence contexts was the main trend induced by infection in soybean roots [54]. In total, 703 and 1,346 genes were found to be hyper- and hypomethylated, respectively. This differential methylation has various effects on gene expression. These vast changes in DNA methylation occur only in the susceptible soybean lines [55]. Probably, permanent changes in DNA methylation play a role in soybean-SCN interaction.
2.2.4. Parasitic plants
Parasitic plants use specialized organs, haustoria, to penetrate the host plant tissues and extract nutrients and water for their growth and reproduction. Besides nutrients and water, haustoria serve as channels for the transports of signaling molecules, protein, DNA, and RNA [56]. About 45% (9518) of the expressed Arabidopsis transcripts were detected in Cuscuta stem parts free of the host tissue, whereas host stem parts that were free of the parasite tissue contained 0.6% of Cuscuta transcripts [57]. A similar pattern was found in the tomato-Cuscuta association, though somewhat lower rates of transfer were estimated to occur. The volumes of mRNA traffic between Cuscuta and the two hosts were consistent in both directions, suggesting that haustorial selectivity is regulated by the host plant. Probably, it reflects some active mechanisms to resist infection present in tomato, such as the secretion of defensive compounds at the infection site [58]. Host mRNAs mostly disappeared from Cuscuta within several hours, but some of them were detected over long distances in parasite stems up to ~20 cm from the haustorial connection [59]. Whether mobile mRNAs have a function remains unclear, though their delayed degradation in foreign tissues suggests some functional significance.
Some sRNAs that move between parasite and host plants are known to function trans-specifically [60]. Relative to the parasite stem, 76 C. campestris sRNA species were significantly upregulated in the host-parasite interface, including 43 miRNAs [61]. Six Arabidopsis mRNAs were predicted to be targets of movable Cuscuta miRNA. No endogenous C. campestris mRNA targets were found to any of the induced miRNAs, suggesting that these miRNAs have evolved to avoid targeting the C. campestris own transcripts. Instead, these miRNAs may function to target mRNAs of the host plant. Indeed, five of these putative target mRNAs were significantly downregulated in parasitized compared with control stems. SEOR1 and AFB3 mRNAs were among the six putative targets of mobile Cuscuta miRNA. Mutant plants seor1 and afb3 showed significantly increased susceptibility to C. campestris. Therefore, the mobile miRNA of C. campestris targets host mRNAs in a biologically relevant way to counteract the host defensive mechanism. Overall, the results suggest that C. campestris trans-species miRNAs function to change host gene expression in a way beneficial to the parasite. The data described collectively indicates that epigenetic interactions shape the dynamics of the “arms race” between host and parasite plants.
3. Short-term epigenetic memory (priming) of stress
- Short-term epigenetic memory (priming) of stress
In natural environments, plants repeatedly experience unfavorable encounters. In evolution, they elaborate specific adaptive mechanisms to overcome various kinds of environmental stress and retain the stress response information for some time after the stress encounters are over. It was shown that stress factors induce alterations in the epigenetic status of stress‐response genes that could still be present for some time after recovery or even in the progeny [62][21,31,36,38,62]. This kind of information used by plants to respond faster or stronger at repeated exposure to the same stress was named stress priming or stress memory [63][36,63]. Arabidopsis plants subjected to several dehydration/water recovery cycles retained more water than plants experiencing dehydration stress for the first time [36]. Moreover, these treatments affected gene expression in two different ways. Some genes were expressed at similar levels during each stress treatment, while other genes significantly increased their expression at repeated treatments relative to the first treatment. Accordingly, genes in the second category could be referred to as “stress memory genes,” while the genes in the first category are just stress-responsive (“non-memory”) genes. Two distinct marks were found at the memory genes during the recovery period: high levels of H3K4me3 and stalled form of RNA polymerase II – Ser5P PolII (phosphorylated at the serine 5th). In contrast, on stress-responsive non-memory genes, these two marks dynamically increased at stress treatment and then decreased to basal levels during the recovery period. At the memory genes, H3K4me3 and Ser5P Pol II persisted for as long as the transcriptional memory lasted. A comprehensive analysis of the Arabidopsis transcriptomes prior to dehydration stress and after the first and third stress exposures revealed a high diversity of memory-type responses [37].
4. Transgenerationally inherited epigenetic memory of stress
- Transgenerationally inherited epigenetic memory of stress
In addition to the memory during priming, epigenetic changes could transmit between plant generations. Multiple cases of such transgenerationally inheritable epigenetic changes (epimutations), both naturally arisen and artificially induced, were described [64]. The formation of stress‐induced transgenerational memory obviously should benefit the plant progeny to achieve a better stress resistance [65][66][65,66].
One of the most widely accepted roles of DNA methylation in plant genomes is the control of transposon activity [67][2,67]. In Arabidopsis, HS transiently destabilized TGS at the constitutive heterochromatin loci rich in transposable elements [68]. At 24 h after HS, TGS re-establishes with a notable exception of a small retrotransposon family ONSEN (Japanese “hot spring”), which retains a high level of transcription. ONSEN transcripts are detected in HS plants directly after the stress treatment and for up to 3 days of recovery. During the recovery period, ONSEN transcripts gradually decrease and become undetectable after 10 days. In the DNA of Arabidopsis plants subjected to HS, a significant increase in the ONSEN copy number was detected. These copy numbers gradually decrease during the 20–30 day recovery period, eventually returning to the initial number of the Columbia accession. No chromosomal integration events are observed in stressed wild-type plants. Surprisingly, frequent transpositions occur in the progeny of RdDM deficient mutant plants subjected to HS. The patterns of new ONSEN insertions are quite variable even between the sibling progeny of the same plant. In the second generation of mutant HS-treated plants, two loci harboring new ONSEN insertions showed heat responsiveness absent in wild-type plants. Therefore, after an induced burst of ONSEN transposition, different subsets of genes in various progeny plants acquire new regulatory properties. Probably, even a transitory relaxation of epigenetic control induced by stress factors could lead to genetic and epigenetic variability, potentially increasing the chances of new adaptive phenotypes.
5. Conclusions
- Conclusions
Many studies showed that epigenetic variation could be an important mechanism to adapt to different habitats. Immobile plants succeed in surviving in a changeable and often hostile environment due to high phenotypic plasticity. Plants have evolved sophisticated mechanisms to respond and adapt to various stresses. These mechanisms operate at various time scales, from short-term physiological and metabolic responses to long-term genetic and epigenetic modifications (Figure 1). The short-term mechanisms are essential in the immediate survival of the stressful conditions, while the long-term modifications could be of evolutionary significance in providing a stable molecular basis for phenotypic plasticity to select for a progeny that is more adapted to a permanently changing environment.
Fundamental features of epigenetic signals, such as their important role in the control of gene expression and their stability and heritability in successive plant generations, support the view of epigenetic variations as a unique adaptation mechanism of evolutionary significance [69][64,69]. Unlike classic mutations, adaptive epigenetic changes can occur at much shorter timescales than those needed to select adaptive mutations. Despite their stability, epigenetic signals are essentially reversible. On the evolutionary time scales, the epigenetically induced adaptive phenotypes would probably be genetically assimilated. However, even considering its short lifetime, epigenetic variability plays an essential role in plant adaptation to a changing environment.
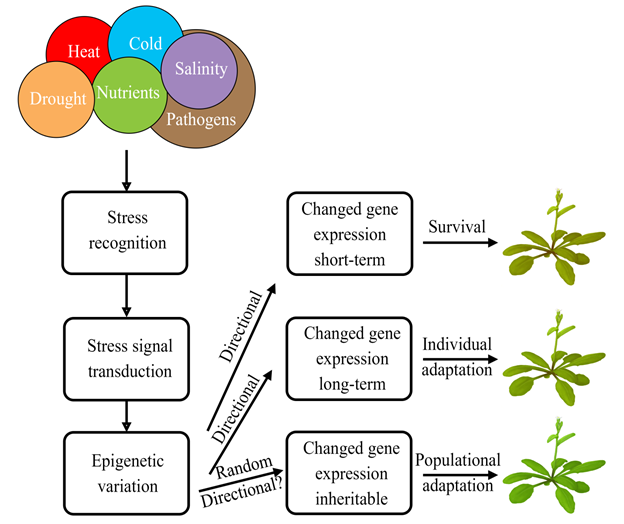
Figure 1. Epigenetic mechanisms of the short-term and long-term plant adaptation to environmental stresses.
Because of their role in regulating gene expression, epigenetic variations can create the phenotypic differences affecting individual fitness and, therefore, can serve as the material of natural selection. Unlike classic genetic mutations, changes in DNA methylation (epimutations) may occur very rapidly in response to environmental stress and provide potential means to cope with it on a very short time scale. Therefore, DNA methylation could be an evolutionary relevant process even in the absence of transgenerational inheritance. However, concerning long-term adaptation and evolution, the most promising epialleles are those capable of being inherited between generations independent of specific genetic loci. Alternative epialleles established in response to environmental stress could give the plants that possess them an alternative phenotype. The frequency of potentially favorable epialleles will be higher in subsequent generations. Even this increased phenotypic plasticity per se could be advantageous in rapidly changing environments since it may help adapt to new environments and provide a more expansive habitat. Based on their dependence on genetic factors, epigenetic variants (epialleles) are classified into three types: (i) “obligate” that display a complete dependency on a genetic feature, (ii) “facilitated” that arise as a result of a genetic feature but could be maintained further in its absence, and (iii) “pure” that do not depend on any genetic features [70]. Most of the epialleles known today are obligate or facilitated, but a few pure epialleles were also found. Thus, significant phenotypic variation can arise due to DNA methylation changes that can potentially be inherited by subsequent generations. However, the epigenetic variation depends, to a very significant extent, on the underlying genetic variation, and these two types of variation should be analyzed together.[1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70]
References
- Feng, S.; Jacobsen, S.E. Epigenetic modifications in plants: an evolutionary perspective. Curr. Opin. Plant Biol. 2011, 14, 179–186, doi:10.1016/j.pbi.2010.12.002.
- Vanyushin, B.F.; Ashapkin, V.V. DNA methylation in higher plants: Past, present and future. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 2011, 1809, 360–368, doi:10.1016/j.bbagrm.2011.04.006.
- Woo, H.R.; Dittmer, T.A.; Richards, E.J. Three SRA-Domain Methylcytosine-Binding Proteins Cooperate to Maintain Global CpG Methylation and Epigenetic Silencing in Arabidopsis. PLoS Genet. 2008, 4, e1000156, doi:10.1371/journal.pgen.1000156.
- Du, J.; Zhong, X.; Bernatavichute, Y.V.; Stroud, H.; Feng, S.; Caro, E.; Vashisht, A.A.; Terragni, J.; Chin, H.G.; Tu, A.; et al. Dual Binding of Chromomethylase Domains to H3K9me2-Containing Nucleosomes Directs DNA Methylation in Plants. Cell 2012, 151, 167–180, doi:10.1016/j.cell.2012.07.034.
- Cao, X.; Jacobsen, S.E. Role of the Arabidopsis DRM Methyltransferases in De Novo DNA Methylation and Gene Silencing. Curr. Biol. 2002, 12, 1138–1144, doi:10.1016/S0960-9822(02)00925-9.
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72, doi:10.1038/nsmb.2735.
- Zemach, A.; Kim, M.Y.; Hsieh, P.-H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis Nucleosome Remodeler DDM1 Allows DNA Methyltransferases to Access H1-Containing Heterochromatin. Cell 2013, 153, 193–205, doi:10.1016/j.cell.2013.02.033.
- Matzke, M.A.; Kanno, T.; Matzke, A.J.M. RNA-Directed DNA Methylation: The Evolution of a Complex Epigenetic Pathway in Flowering Plants. Annu. Rev. Plant Biol. 2015, 66, 243–267, doi:10.1146/annurev-arplant-043014-114633.
- Wendte, J.M.; Pikaard, C.S. The RNAs of RNA-directed DNA methylation. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 2017, 1860, 140–148, doi:10.1016/j.bbagrm.2016.08.004.
- Gehring, M.; Reik, W.; Henikoff, S. DNA demethylation by DNA repair. Trends Genet. 2009, 25, 82–90, doi:10.1016/j.tig.2008.12.001.
- Tang, K.; Lang, Z.; Zhang, H.; Zhu, J.-K. The DNA demethylase ROS1 targets genomic regions with distinct chromatin modifications. Nat. Plants 2016, 2, 16169, doi:10.1038/nplants.2016.169.
- Berr, A.; Shafiq, S.; Shen, W.-H. Histone modifications in transcriptional activation during plant development. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 2011, 1809, 567–576, doi:10.1016/j.bbagrm.2011.07.001.
- Zhang, X.; Bernatavichute, Y.V.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009, 10, R62, doi:10.1186/gb-2009-10-6-r62.
- Zhang, X.; Clarenz, O.; Cokus, S.; Bernatavichute, Y.V.; Pellegrini, M.; Goodrich, J.; Jacobsen, S.E. Whole-Genome Analysis of Histone H3 Lysine 27 Trimethylation in Arabidopsis. PLoS Biol. 2007, 5, e129, doi:10.1371/journal.pbio.0050129.
- Zhang, X.; Germann, S.; Blus, B.J.; Khorasanizadeh, S.; Gaudin, V.; Jacobsen, S.E. The Arabidopsis LHP1 protein colocalizes with histone H3 Lys27 trimethylation. Nat. Struct. Mol. Biol. 2007, 14, 869–871, doi:10.1038/nsmb1283.
- Bernatavichute, Y.V.; Zhang, X.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-Wide Association of Histone H3 Lysine Nine Methylation with CHG DNA Methylation in Arabidopsis thaliana. PLoS ONE 2008, 3, e3156, doi:10.1371/journal.pone.0003156.
- Lee, C.H.; Carroll, B.J. Evolution and Diversification of Small RNA Pathways in Flowering Plants. Plant Cell Physiol. 2018, doi:10.1093/pcp/pcy167.
- Verhoeven, K.J.F.; Jansen, J.J.; van Dijk, P.J.; Biere, A. Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytol. 2010, 185, 1108–1118, doi:10.1111/j.1469-8137.2009.03121.x.
- Zhu, J.-K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324, doi:10.1016/j.cell.2016.08.029.
- Yang, R.; Hong, Y.; Ren, Z.; Tang, K.; Zhang, H.; Zhu, J.-K.; Zhao, C. A Role for PICKLE in the Regulation of Cold and Salt Stress Tolerance in Arabidopsis. Front. Plant Sci. 2019, 10, 900, doi:10.3389/fpls.2019.00900.
- Yang, R.; Zheng, Z.; Chen, Q.; Yang, L.; Huang, H.; Miki, D.; Wu, W.; Zeng, L.; Liu, J.; Zhou, J.-X.; et al. The developmental regulator PKL is required to maintain correct DNA methylation patterns at RNA-directed DNA methylation loci. Genome Biol. 2017, 18, 103, doi:10.1186/s13059-017-1226-y.
- Carter, B.; Bishop, B.; Ho, K.K.; Huang, R.; Jia, W.; Zhang, H.; Pascuzzi, P.E.; Deal, R.B.; Ogas, J. The Chromatin Remodelers PKL and PIE1 Act in an Epigenetic Pathway That Determines H3K27me3 Homeostasis in Arabidopsis. Plant Cell 2018, 30, 1337–1352, doi:10.1105/tpc.17.00867.
- Park, J.; Lim, C.J.; Shen, M.; Park, H.J.; Cha, J.-Y.; Iniesto, E.; Rubio, V.; Mengiste, T.; Zhu, J.-K.; Bressan, R.A.; et al. Epigenetic switch from repressive to permissive chromatin in response to cold stress. Proc. Natl. Acad. Sci. 2018, 115, E5400–E5409, doi:10.1073/pnas.1721241115.
- Ohama, N.; Sato, H.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Transcriptional Regulatory Network of Plant Heat Stress Response. Trends Plant Sci. 2017, 22, 53–65, doi:10.1016/j.tplants.2016.08.015.
- Popova, O.V.; Dinh, H.Q.; Aufsatz, W.; Jonak, C. The RdDM Pathway Is Required for Basal Heat Tolerance in Arabidopsis. Mol. Plant 2013, 6, 396–410, doi:10.1093/mp/sst023.
- Lämke, J.; Brzezinka, K.; Altmann, S.; Bäurle, I. A hit‐and‐run heat shock factor governs sustained histone methylation and transcriptional stress memory. EMBO J. 2016, 35, 162–175, doi:10.15252/embj.201592593.
- Ferreira, L.J.; Azevedo, V.; Maroco, J.; Oliveira, M.M.; Santos, A.P. Salt Tolerant and Sensitive Rice Varieties Display Differential Methylome Flexibility under Salt Stress. PLOS ONE 2015, 10, e0124060, doi:10.1371/journal.pone.0124060.
- Wang, W.; Huang, F.; Qin, Q.; Zhao, X.; Li, Z.; Fu, B. Comparative analysis of DNA methylation changes in two rice genotypes under salt stress and subsequent recovery. Biochem. Biophys. Res. Commun. 2015, 465, 790–796, doi:10.1016/j.bbrc.2015.08.089.
- Baek, D.; Jiang, J.; Chung, J.-S.; Wang, B.; Chen, J.; Xin, Z.; Shi, H. Regulated AtHKT1 Gene Expression by a Distal Enhancer Element and DNA Methylation in the Promoter Plays an Important Role in Salt Tolerance. Plant Cell Physiol. 2011, 52, 149–161, doi:10.1093/pcp/pcq182.
- Kim, J.-M.; Sasaki, T.; Ueda, M.; Sako, K.; Seki, M. Chromatin changes in response to drought, salinity, heat, and cold stresses in plants. Front. Plant Sci. 2015, 6, doi:10.3389/fpls.2015.00114.
- Sani, E.; Herzyk, P.; Perrella, G.; Colot, V.; Amtmann, A. Hyperosmotic priming of Arabidopsis seedlings establishes a long-term somatic memory accompanied by specific changes of the epigenome. Genome Biol. 2013, 14, R59, doi:10.1186/gb-2013-14-6-r59.
- Xu, R.; Wang, Y.; Zheng, H.; Lu, W.; Wu, C.; Huang, J.; Yan, K.; Yang, G.; Zheng, C. Salt-induced transcription factor MYB74 is regulated by the RNA-directed DNA methylation pathway in Arabidopsis. J. Exp. Bot. 2015, 66, 5997–6008, doi:10.1093/jxb/erv312.
- Avramova, Z. Transcriptional ‘memory’ of a stress: transient chromatin and memory (epigenetic) marks at stress-response genes. Plant J. 2015, 83, 149–159, doi:10.1111/tpj.12832.
- Avramova, Z. Defence-related priming and responses to recurring drought: Two manifestations of plant transcriptional memory mediated by the ABA and JA signalling pathways: dehydration stress memory and jasmonic acid priming. Plant Cell Environ. 2019, 42, 983–997, doi:10.1111/pce.13458.
- Ding, Y.; Avramova, Z.; Fromm, M. The Arabidopsis trithorax-like factor ATX1 functions in dehydration stress responses via ABA-dependent and ABA-independent pathways: ATX1 functions in dehydration stress responses. Plant J. 2011, 66, 735–744, doi:10.1111/j.1365-313X.2011.04534.x.
- Ding, Y.; Fromm, M.; Avramova, Z. Multiple exposures to drought “train” transcriptional responses in Arabidopsis. Nat. Commun. 2012, 3, 740, doi:10.1038/ncomms1732.
- Ding, Y.; Liu, N.; Virlouvet, L.; Riethoven, J.-J.; Fromm, M.; Avramova, Z. Four distinct types of dehydration stress memory genes in Arabidopsis thaliana. BMC Plant Biol. 2013, 13, 229, doi:10.1186/1471-2229-13-229.
- Virlouvet, L.; Fromm, M. Physiological and transcriptional memory in guard cells during repetitive dehydration stress. New Phytol. 2015, 205, 596–607, doi:10.1111/nph.13080.
- Gupta, A.; Rico-Medina, A.; Caño-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269, doi:10.1126/science.aaz7614.
- Ramirez-Prado, J.S.; Abulfaraj, A.A.; Rayapuram, N.; Benhamed, M.; Hirt, H. Plant Immunity: From Signaling to Epigenetic Control of Defense. Trends Plant Sci. 2018, 23, 833–844, doi:10.1016/j.tplants.2018.06.004.
- Zheng, X.; Chen, L.; Li, M.; Lou, Q.; Xia, H.; Wang, P.; Li, T.; Liu, H.; Luo, L. Transgenerational Variations in DNA Methylation Induced by Drought Stress in Two Rice Varieties with Distinguished Difference to Drought Resistance. PLoS ONE 2013, 8, e80253, doi:10.1371/journal.pone.0080253.
- Secco, D.; Whelan, J.; Rouached, H.; Lister, R. Nutrient stress-induced chromatin changes in plants. Curr. Opin. Plant Biol. 2017, 39, 1–7, doi:10.1016/j.pbi.2017.04.001.
- Widiez, T.; El Kafafi, E.S.; Girin, T.; Berr, A.; Ruffel, S.; Krouk, G.; Vayssières, A.; Shen, W.-H.; Coruzzi, G.M.; Gojon, A.; et al. HIGH NITROGEN INSENSITIVE 9 (HNI9)-mediated systemic repression of root NO 3 − uptake is associated with changes in histone methylation. Proc. Natl. Acad. Sci. 2011, 108, 13329–13334, doi:10.1073/pnas.1017863108.
- Fan, H.; Zhang, Z.; Wang, N.; Cui, Y.; Sun, H.; Liu, Y.; Wu, H.; Zheng, S.; Bao, S.; Ling, H.-Q. SKB1/PRMT5-mediated histone H4R3 dimethylation of Ib subgroup bHLH genes negatively regulates iron homeostasis in Arabidopsis thaliana. Plant J. 2014, 77, 209–221, doi:10.1111/tpj.12380.
- Xing, J.; Wang, T.; Liu, Z.; Xu, J.; Yao, Y.; Hu, Z.; Peng, H.; Xin, M.; Yu, F.; Zhou, D.; et al. GENERAL CONTROL NONREPRESSED PROTEIN5-Mediated Histone Acetylation of FERRIC REDUCTASE DEFECTIVE3 Contributes to Iron Homeostasis in Arabidopsis. Plant Physiol. 2015, 168, 1309–1320, doi:10.1104/pp.15.00397.
- Chandrika, N.N.P.; Sundaravelpandian, K.; Yu, S.-M.; Schmidt, W. ALFIN-LIKE 6 is involved in root hair elongation during phosphate deficiency in Arabidopsis. New Phytol. 2013, 198, 709–720, doi:10.1111/nph.12194.
- Secco, D.; Wang, C.; Shou, H.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife 2015, 4, e09343, doi:10.7554/eLife.09343.
- Yong-Villalobos, L.; González-Morales, S.I.; Wrobel, K.; Gutiérrez-Alanis, D.; Cervantes-Peréz, S.A.; Hayano-Kanashiro, C.; Oropeza-Aburto, A.; Cruz-Ramírez, A.; Martínez, O.; Herrera-Estrella, L. Methylome analysis reveals an important role for epigenetic changes in the regulation of the Arabidopsis response to phosphate starvation. Proc. Natl. Acad. Sci. 2015, 112, E7293–E7302, doi:10.1073/pnas.1522301112.
- Martínez-Pérez, M.; Aparicio, F.; López-Gresa, M.P.; Bellés, J.M.; Sánchez-Navarro, J.A.; Pallás, V. Arabidopsis m 6 A demethylase activity modulates viral infection of a plant virus and the m 6 A abundance in its genomic RNAs. Proc. Natl. Acad. Sci. 2017, 114, 10755–10760, doi:10.1073/pnas.1703139114.
- Deleris, A.; Halter, T.; Navarro, L. DNA Methylation and Demethylation in Plant Immunity. Annu. Rev. Phytopathol. 2016, 54, 579–603, doi:10.1146/annurev-phyto-080615-100308.
- Yu, A.; Lepere, G.; Jay, F.; Wang, J.; Bapaume, L.; Wang, Y.; Abraham, A.-L.; Penterman, J.; Fischer, R.L.; Voinnet, O.; et al. Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. 2013, 110, 2389–2394, doi:10.1073/pnas.1211757110.
- López Sánchez, A.; Stassen, J.H.M.; Furci, L.; Smith, L.M.; Ton, J. The role of DNA (de)methylation in immune responsiveness of Arabidopsis. Plant J. 2016, 88, 361–374, doi:10.1111/tpj.13252.
- Meller, B.; Kuźnicki, D.; Arasimowicz-Jelonek, M.; Deckert, J.; Floryszak-Wieczorek, J. BABA-Primed Histone Modifications in Potato for Intergenerational Resistance to Phytophthora infestans. Front. Plant Sci. 2018, 9, 1228, doi:10.3389/fpls.2018.01228.
- Rambani, A.; Rice, J.H.; Liu, J.; Lane, T.; Ranjan, P.; Mazarei, M.; Pantalone, V.; Stewart, C.N.; Staton, M.; Hewezi, T. The Methylome of Soybean Roots during the Compatible Interaction with the Soybean Cyst Nematode. Plant Physiol. 2015, 168, 1364–1377, doi:10.1104/pp.15.00826.
- Rambani, A.; Pantalone, V.; Yang, S.; Rice, J.H.; Song, Q.; Mazarei, M.; Arelli, P.R.; Meksem, K.; Stewart, C.N.; Hewezi, T. Identification of introduced and stably inherited DNA methylation variants in soybean associated with soybean cyst nematode parasitism. New Phytol. 2020, 227, 168–184, doi:10.1111/nph.16511.
- Gaut, B.S.; Miller, A.J.; Seymour, D.K. Living with Two Genomes: Grafting and Its Implications for Plant Genome-to-Genome Interactions, Phenotypic Variation, and Evolution. Annu. Rev. Genet. 2019, 53, 195–215, doi:10.1146/annurev-genet-112618-043545.
- Kim, G.; LeBlanc, M.L.; Wafula, E.K.; dePamphilis, C.W.; Westwood, J.H. Genomic-scale exchange of mRNA between a parasitic plant and its hosts. Science 2014, 345, 808–811, doi:10.1126/science.1253122.
- Kaiser, B.; Vogg, G.; Fürst, U.B.; Albert, M. Parasitic plants of the genus Cuscuta and their interaction with susceptible and resistant host plants. Front. Plant Sci. 2015, 6, doi:10.3389/fpls.2015.00045.
- LeBlanc, M.; Kim, G.; Patel, B.; Stromberg, V.; Westwood, J. Quantification of tomato and Arabidopsis mobile RNAs trafficking into the parasitic plant Cuscuta pentagona. New Phytol. 2013, 200, 1225–1233, doi:10.1111/nph.12439.
- Alakonya, A.; Kumar, R.; Koenig, D.; Kimura, S.; Townsley, B.; Runo, S.; Garces, H.M.; Kang, J.; Yanez, A.; David-Schwartz, R.; et al. Interspecific RNA Interference of SHOOT MERISTEMLESS-Like Disrupts Cuscuta pentagona Plant Parasitism. Plant Cell 2012, 24, 3153–3166, doi:10.1105/tpc.112.099994.
- Shahid, S.; Kim, G.; Johnson, N.R.; Wafula, E.; Wang, F.; Coruh, C.; Bernal-Galeano, V.; Phifer, T.; dePamphilis, C.W.; Westwood, J.H.; et al. MicroRNAs from the parasitic plant Cuscuta campestris target host messenger RNAs. Nature 2018, 553, 82–85, doi:10.1038/nature25027.
- Sanchez, D.H.; Paszkowski, J. Heat-Induced Release of Epigenetic Silencing Reveals the Concealed Role of an Imprinted Plant Gene. PLoS Genet. 2014, 10, e1004806, doi:10.1371/journal.pgen.1004806.
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124, doi:10.1186/s13059-017-1263-6.
- Ashapkin, V.V.; Kutueva, L.I.; Vanyushin, B.F. Epigenetic variability in plants: Heritability, adaptability, evolutionary significance. Russ. J. Plant Physiol. 2016, 63, 181–192, doi:10.1134/S1021443716020059.
- Cong, W.; Miao, Y.; Xu, L.; Zhang, Y.; Yuan, C.; Wang, J.; Zhuang, T.; Lin, X.; Jiang, L.; Wang, N.; et al. Transgenerational memory of gene expression changes induced by heavy metal stress in rice (Oryza sativa L.). BMC Plant Biol. 2019, 19, 282, doi:10.1186/s12870-019-1887-7.
- Zheng, X.; Chen, L.; Xia, H.; Wei, H.; Lou, Q.; Li, M.; Li, T.; Luo, L. Transgenerational epimutations induced by multi-generation drought imposition mediate rice plant’s adaptation to drought condition. Sci. Rep. 2017, 7, 39843, doi:10.1038/srep39843.
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220, doi:10.1038/nrg2719.
- Ito, H.; Gaubert, H.; Bucher, E.; Mirouze, M.; Vaillant, I.; Paszkowski, J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 2011, 472, 115–119, doi:10.1038/nature09861.
- Eichten, S.R.; Schmitz, R.J.; Springer, N.M. Epigenetics: Beyond Chromatin Modifications and Complex Genetic Regulation. Plant Physiol. 2014, 165, 933–947, doi:10.1104/pp.113.234211.
- Schmitz, R.J.; Ecker, J.R. Epigenetic and epigenomic variation in Arabidopsis thaliana. Trends Plant Sci. 2012, 17, 149–154, doi:10.1016/j.tplants.2012.01.001.