Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by F. David Rodriguez and Version 2 by Catherine Yang.
Substance P (SP), the first isolated neuropeptide, belongs to the family of tachykinin peptides and is the natural ligand of neurokinin-1 receptors (NK-1R), also named SP receptors. The undecapeptide activates the receptor after specifically binding to the protein and triggers intracellular signals leading to different biochemical events and subsequent physiological responses.
- substance P
- neurokinin-1 receptors (NK-1R)
- NK-1R antagonists
1. G Protein-Coupled Receptors (GPCRs)
G protein-coupled receptors (GPCRs) are abundant and ubiquitous membrane proteins that respond to stimuli from photons to ions, small organic compounds, peptides, and macromolecules. They are responsible for multiple cellular responses and share a serpentine architecture with a package of seven transmembrane helices, alternate intracellular and extracellular loops, the C-terminus lying in the cytoplasm, and the N-terminus expanding towards the extracellular space [1][2][3][4][5][6][7][17,57,58,59,60,61,62]. GPCRs activate different G protein and β-arrestin-dependent mechanisms that control many physiological and pathological outcomes [8][63] related to immune responses, mood, cognition, blood pressure control, pain control, taste, vision, and olfaction [6][9][10][11][61,64,65,66]. Signaling can also occur via endosomal membranes that facilitate the activation of localized signals [12][13][14][67,68,69]. The group is subdivided into classes considering their sequences’ similarities and structural features: A (rhodopsin), B (secretin), B2 (adhesion), C (glutamate receptors), D1 (Ste2-like fungal pheromone), family F (frizzled, FZD, and smoothened, SMO), and T (taste) [10][15][16,65]. Extensive and meticulous structural studies of GPCRs provide essential information that allows for a better understanding of their function. They also improved drug design based on the knowledge of protein conformational movements linked to activation and inactivation states and biased agonist signaling [2][10][16][17][18][57,65,70,71,72]. X-ray crystallography studies, molecular dynamics simulations, and the mutational analysis of allosteric interfaces with hydrogen bonding networks in transmembrane helices determine states of activity that trigger intracellular signaling [6][7][19][20][21][61,62,73,74,75]. Updated information on the structural studies of GPCRs can be found in the GPCRs database [1][15][16,17]. GPCRs serve as a target for pharmacological intervention. Approximately 35% of officially approved drugs target GPCRs directly or GPCR-associated proteins [22][23][76,77]. The list may grow more prominent due to new methods of drug design exploration based on novel receptor regulation concepts, allosteric changes, biased signaling, and receptor heteromerization [24][25][26][27][78,79,80,81]. Additionally, post-translational modifications (phosphorylation, glycosylation, ubiquitination, and palmitoylation) influence receptor folding, dimerization, and function [28][82]. Soundly, these modifications should also be focused on when studying the pharmacological manipulation of GPCRs.
Class A GPCRs (to which NK-1 receptors belong) are the most abundant and studied. They have an enormous influence on human physiology and pathology. Agonist binding to these receptors activates G protein recruitment by moving helix number 6 towards the exterior of the receptor bundle caused by a common activation mechanism affecting specific residues [18][72]. The first structures defined with X-ray diffraction data were bovine rhodopsin [29][83] and human β-adrenergic receptors [12][30][67,84]. More than a hundred structures of GPCRs have been resolved to date. Conserved sequence motifs, such as D(E)R3.50Y in TM3 and D/E6.30 in TM6, are responsible for the inactivation state of the receptors by establishing an ionic bridge tightening TM3 and TM6 together, thus preventing the flexible outward movement of TM6 necessary for activation and signaling.
2. NK-1R Signaling Pathways
The NK-1 receptor belongs to class A of GPCRs. Upon activation, it signals through different pathways depending on its intrinsic performance, topology within the membrane bilayer, tissue localization, and the type of ligand. Agonist ligands of NK-1R include SP, SP analogs, and neurokinin A (NKA) [31][32][33][34][85,86,87,88]. The main signaling routes activate Gq and Gs heterotrimeric proteins, causing intracellular variations in second messenger concentrations, such as calcium, cAMP, or IP3 [35][36][89,90]. Figure 17 depicts the main signaling pathways activated by NK-1 receptors, leading to operative protein phosphorylation cascades and metabolic regulations responsible for physiological and pathological consequences.
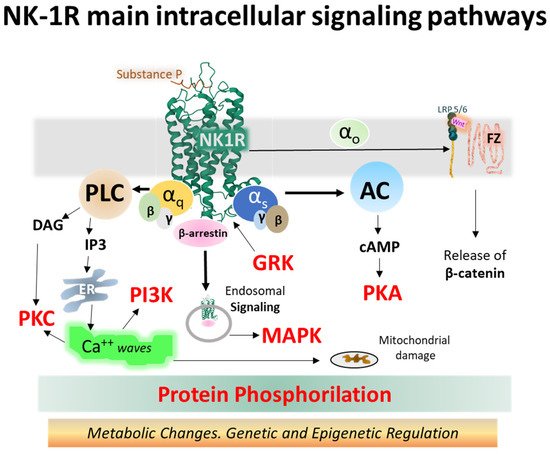
Figure 17. Intracellular NK-1R signaling pathways turned on after binding SP and activating different alpha subunits (Gq and Gs, mainly) and beta-gamma dimers of heterotrimeric G proteins (the structure of NK-1R in green is from the Protein Data Bank [37][30], PDB Id. 2KS9) [38][31]. Abbreviations: AC (adenylyl cyclase), cAMP (cyclic 3′-5′ adenosine monophosphate), DAG (diacylglycerol), ER (endoplasmic reticulum), FZ (frizzled), GRK (G protein-coupled receptor kinase), IP3 (inositol 1,4,5 trisphosphate), LRP 5/6 (low-density lipoprotein receptor-related protein), MAPK (mitogen-activated protein kinase), NK-1R (neurokinin-1 receptor), PI3K (phosphatidylinositol 3-kinase), PKA (protein kinase A), PKC (protein kinase C), PLC (phospholipase C), and Wnt (wingless-related integration site) [35][39][40][41][42][43][44][45][46][47][48][26,89,91,92,93,94,95,96,97,98,99].
The observed differential activation of NK-1R signaling cascades by agonists may be caused by structural coupling between the receptor and agonist, where efficacy and selectivity lie in different architectural settings. The interface formed by amino acid positions E2.50, S3.39, and N7.49 sets in a net of hydrogen bonds (Figure 5) and facilitates receptor conformations leading to biased receptor activation (Gs, Gq, and β-arrestin) [49][55]. In this hydrogen bond network built by the amino acid lateral chains, the alanine substitution of E2.50 abolished SP-activated Gq signaling measured by the accumulation of inositol 1, 4,5 trisphosphate (IP3). However, the alanine substitution of N7.49 and S3.39 had a marginal effect on Gq stimulation.
SP and neurokinin A (NKA) and neurokinin B (NKB) bind NK-1 receptors; however, differences in the charge distribution and amino acid composition of the peptides and their interaction with the anionic surface formed by acidic residues in extracellular loops 2 and 3 favor SP binding (higher affinity) over the other two tachykinins [50][45]. The structural resolution of NK-1 receptors bound to Gs and Gq proteins contributed to determining that the receptor similarly binds to both G proteins [50][45]. However, the binding to Gq is slightly favored and accords with the observed preference of Gq over Gs signaling [51][100]. Mutagenesis experiments and the structural dynamics analysis of ligand–receptor interactions [31][32][52][47,85,86] showed that SP and its truncated form SP6-11 (lacking the N-terminal region of the undecapeptide) triggered calcium signaling with similar potency when bound to wild-type NK-1 receptors. However, NKA and SP6-11 induce a weaker cAMP accumulation response than SP. Additionally, experiments with NK-1R bearing conservative mutations of amino acids establishing hydrogen bonding nets deeper inside the pocket (N852.57, N892.61, H1083.28, and Y2877.35) with the C-terminal region of SP (Figure 28) did not affect the potency and maximal response of the undecapeptide. In contrast, truncated SP6-11-dependent responses significantly decreased [52][47].
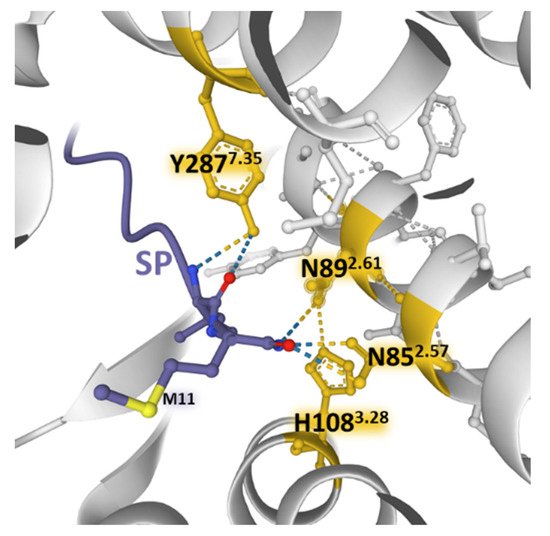
Figure 28. Weak bonding interactions of different amino acid residues (yellow) coming into contact with the C-terminal region of SP (blue) deep inside the orthosteric pocket of the NK-1 receptor (gray) (PDB Id: 7RMG [52][47]). Conservative mutations of the depicted positions do not affect the SP-induced increase in intracellular Ca2+ concentrations. The figure also indicates residue M11 of the undecapeptide. The structure is from the Protein Data Bank [53][41], drawn with the Mol* software [54][43].
The findings point out that the calcium signaling depended on the interaction of the C-terminal region of SP with the receptor. However, the findings also showed that contact between the central and N-terminal parts of SP with the extracellular domains of NK-1R (ECL2 and N-terminus area) is essential for signaling through Gs and not only for ligand recognition and selectivity. Molecular dynamics simulations reported that the residues F7 and M11 in SP6-11-truncated peptide adopt more orientations than F7 and M11 in SP undecapeptide and establish differential contacts with TM7 and TM6 of the NK-1R [52][47]. It is worth noting that preferential Gq activation obtained with SP6-11 reinforces the assumption that connections between the N-terminal region of SP with the exterior of NK-1R determined receptor activation-dependent signaling.
Other GPCRs also exhibit biased agonism responsible for activating singular intracellular metabolic reactions, producing a signaling signature at a given time and in a particular target cell [20][55][74,101].
The truncated NK-1 receptor isoform lacks the intracellular C-terminal region [56][57][21,102]. Full (NK-1R-F) and truncated NK-1 (NK-1R-T) receptor isoforms display functional differences affecting intracellular signaling [39][26]. The exclusion of the C-terminal tail impairs receptor internalization, making the protein resistant to desensitization [56][21]. Additionally, the interaction with transducer G proteins is weaker, and calcium release, protein kinase C (PKC) activation, and other phosphorylation events appear delayed [58][59][60][5,25,103].
The so-called canonical Wnt/β-catenin signaling pathways encompass a group of membrane-bound and intracellular proteins, and the route is closely related to the control of immunomodulation and cell proliferation. The activation of the FZ/LRP 5/6 complex (frizzled/low-density lipoprotein receptor-related protein 5/6) by secreted cysteine-rich glycoproteins Wnt (wingless-related integration site) (see Figure 17) mobilizes an effector G protein, disrupts the Axin-GSK3-APC-CK1 complex and impedes the phosphorylation of β-catenin by the kinases GSK-3β and CK1 (casein kinase 1). Consequently, β-catenin accumulates in the cytosol and moves to the nucleus, where it associates with TCFs/LEF (T-cell factor/lymphoid-enhancer-binding factor) proteins and other transcription factors and co-triggers the transcription of target genes by binding to specific promoter sequences [47][61][62][63][64][98,104,105,106,107]. β-catenin may also translocate to the membrane and stabilize it by building cell adhesion complexes [64][65][107,108].
Experimental studies using agonists and antagonists of NK-1R showed that the agonist turning-on of NK-1R induced an increased expression of β-catenin and GSK3β and diminished the expression of DKK1 (Dickkopf 1), an inhibitor of Wnt [66][109]. Additionally, selective antagonists of NK-1 receptors inhibited cell proliferation in several experimental scenes by influencing Wnt/β-catenin pathways [40][65][67][68][91,108,110,111].
The NK-1R system controls numerous intracellular processes, including cell growth, cytoskeletal organization, metabolic reaction, or gene expression through biochemical reactions that contribute to cell homeostasis [69][112]. Malfunction of the receptor induces dysregulation of intracellular signaling cascades, and disease may appear. NK-1R-triggered signaling complexes participating in cell disruption led to inflammation, calcium overloads, altered cell metabolism and growth, and disrupted cell migration conveying to cancer, pain, psychiatric disorders, and other pathologies. Molecular complexes responsible include the Wnt/β-catenin [40][91], β-arrestins scaffolds [69][112], JNK and p38/MAPK [70][113], NF-kB [70][71][113,114], PKCδ/ERK/P65 [72][115], or metalloproteases MMP-2/MMP-9 [73][116], to mention a few (Figure 17).
Aside from fine structural studies revealing activated and inactivated conformations of the receptors induced by movements of receptor domains, determining the binding kinetics of both agonist and antagonist ligands deserves attention to ascertain signal transduction outcomes and determine new drugs’ specificity, efficacy, modulation, and potency [74][117].