The homeostasis of cell volume, which is essential for the survival of all mammalian cells, requires the dynamically regulated transport of ions across the plasmalemma. While the individual “effector” molecules involved in cell volume regulation such as ion channels (e.g., VRACs) and transporters (e.g., the NKCC1 and KCC2 cation-Cl- cotransporters (CCCs) are well established, the “sensor” and “transducer” molecules that coordinate their activities remain poorly characterized. Dysregulation or failure of cell volume homeostasis occurs in numerous clinical contexts, with cerebral edema (i.e., “brain swelling”) – as occurs after stroke or trauma – being a quintessential example. In the review, we summarise previous work and recent advances that attest to cation chloride cotransporters’s growing potential as a therapeutic target for Cardiovascular diseases (CVDs).
- cardiovascular disease
- hypertension
- atherosclerosis
- electroneutral transport
- cation-chloride-cotransporters
- KCCs
- NKCCs
1. Introduction
Cardiovascular diseases (CVDs) are the leading cause of mortality globally. It has been estimated that countries in the European region alone spent GBP 80 billion on health care costs related to CVDs[1][2][3]. CVDs are a group of health complications that affect the heart and circulation, including coronary heart disease, heart attack, stroke, heart failure, arrhythmia, and more. Coronary heart disease (CHD) is mainly caused by reduced or blockage of blood flow to the heart and can lead to heart attacks (blocked blood flow to the heart) and heart failure (failure of the heart to pump blood). CHD could also cause heart valve problems and abnormal rhythms (arrhythmias). A stroke occurs due to blockages of blood flow to the brain. Risk factors associated with CVDs include hypertension, diabetes, atherosclerosis, smoking, obesity and high cholesterol. Among the risk factors, hypertension and atherosclerosis are the main underlying causes of CVD. The renal system plays a critical role in blood pressure homeostasis. Water retention in the kidney, due to increased sodium reabsorption, causes extracellular fluid (ECF) volume expansion [4][4]. The volume expansion contributes to the elevation of preload which contributes to the increase in blood pressure. Hypertension could also lead to atherosclerosis. Atherosclerosis is the process of hardening and narrowing of the blood vessels. Vascular proliferation is one of the contributing factors to the pathophysiology of atherosclerosis. Vascular proliferation is the phenotypic changes of the vascular smooth muscle cells (VSMCs), the most numerous cell type in the blood vessel, characterized by the reversible transition from a quiescent contractile state to a synthetic migratory phenotype[5]. Although there are preventive measures to reduce the burden of CVDs, atherosclerosis is common in people over the age of 65 and 1.3 billion people worldwide are estimated to be hypertensive[1][2][3]. Thus current research aims are to discover novel targets for the treatment of CVDs. Our recent research developed a molecular compound termed ZT-1a for the treatment of stroke, by modulating the activities of cation chloride cotransporters (CCC)[6]. In the review, the focus is to summarise previous work and recent advances that attest to CCC’s growing potential as a therapeutic target for CVDs.
2. Cation Chloride Cotransporter
2.1 Evaluation of the Cation Chloride Cotransporter Family
Comprehensive phylogenetic analysis by Hartmann et al. revealed the existence of a single ancestral cation chloride cotransporter (CCC) gene in the Archean, Methanosarcina acetivorans[7] . The ancestral gene appears to be the base of numerous duplication events which led to paralogous CCC subfamilies in Archean and eukaryotes. These subfamilies consist of sodium chloride cotransporter (NCC), sodium potassium chloride cotransporters (NKCCs, NKCC1 and NKCC2; with NCC collectively referred to as N[K]CCs), potassium chloride cotransporters (KCCs, KCC1–4), polyamine transporter (CCC9) and CCC-interacting protein (CIP1). NCC and NKCCs use Na+ in their stoichiometric translocation of Cl−, whereas KCCs using K+. CIP1 have been shown to inhibit NKCC1 activity[8] and enhance KCC2 [9][9] activity in cultured cells. CCC9 remains unclassified. CCCs provide electroneutral transport of sodium, potassium and chloride across the plasma membrane; with an exception of NCC, all CCCs are inhibited by several structurally similar compounds, such as bumetanide and furosemide (Table 1). It is important to note that KCCs are only weakly inhibited by these compounds. Further gene-loss events resulted in the complex distribution of CCCs across the taxa.
Gene duplication within the CCC subfamilies of vertebrates resulted in subfunctionalization[7]. This process created a number of isoforms with different expression patterns and functionality in various organs and tissues. Further variants are generated by alternative splicing of the isoforms with various ratios of expression in human tissues (Table 1).
Table 1. Major characteristics of Cl−-coupled cation cotransporters [10][11][12][13].
|
Gene |
Human chromosome localization |
Protein |
Transported ions |
Alternative spicing |
Tissue distribution, cellular/subcellular expression |
Link to disease |
Inhibitors, IC50 (μM) |
|
SLC12A2 |
5q23.3 |
NKCC1 |
Na+, K+, Cl− |
Isoforms A and B [14] |
Ubiquitous: basolateral membrane of epithelial cells, non-epithelial cells |
Schizophrenia [15] |
Bumetanide, 0.05–0.60; Furosemide, 10–50; ARN23746[16] Azosemide, 0.246–0.197 |
|
SLC12A1 |
15q21.1 |
NKCC2 |
Na+, K+, Cl− |
Isoforms A, B and F [17] |
Kidney-specific: apical membrane of the thick ascending limb |
Bartter syndrome type I[18] |
Bumetanide, 0.10–0.50; Furosemide, 15–60 |
|
SLC12A3 |
16q13 |
NCC |
Na+, Cl− |
NA |
Kidney-specific: apical membrane of the distal convoluted tubule |
Gitelman syndrome [19] |
Hydrochlorothiazide, 70[20]; Metolazone, 0.3[21] |
|
SLC12A4 |
16q22 |
KCC1 |
K+, Cl− |
NA |
Ubiquitous |
NA |
Bumetanide, 60[22]; Furosemide, 40[22]]; DIOA, ~10 |
|
SLC12A5 |
20q13 |
KCC2 |
K+, Cl− |
NA |
Neurones |
Epilepsy[23] |
Bumetanide, 55; Furosemide, 10; VU 0463271, 0.061[24]; DIOA, ~10 |
|
SLC12A6 |
15q14 |
KCC3 |
K+, Cl− |
Isoforms A and B[25] |
Ubiquitous |
Agenesis of the Corpus Callosum with Peripheral Neuropathy (ACCPN)[26] |
Bumetanide, 40; Furosemide, 25; DIOA,~10 |
|
SLC12A7 |
5p15 |
KCC4 |
K+, Cl− |
NA |
Ubiquitous |
Renal tubular acidosis[27] |
Bumetanide, 900; Furosemide, 900; DIOA, ~10 |
|
SLC12A8 |
3q21 |
CCC9 |
NA |
Six isoforms [28] |
Ubiquitous |
Psoriasis[28] |
NA |
|
SLC12A9 |
7q22 |
CIP1 |
NA |
NA |
Ubiquitous |
NA |
NA |
NA, information not available.
The topology of all CCCs consists of 12 transmembrane domains (TM) domains and large intracellular amino (N) and carboxyl (C) terminal domains[29]. Notably, the KCC and N[K]CC subfamilies vary in their position of the long extracellular loop: between TM5 and TM6 in KCCs and between TM7 and TM8 in the N[K]CC subfamily. Within the isoforms of the subfamilies, the TMs and C-terminus are highly conserved. In contrast, the N terminus is considerably more variable amongst isoforms[29]. Binding of diuretics occurs in the conserved TMs and not the variable termini[29]. Therefore, loop diuretics like furosemide and bumetanide are used to characterize the physiological function of KCCs and NKCCs with little specification for each paralog. The termini are involved in the regulation of transport activity indicative of the sites for post-translational modifications like phosphorylation that are located within them.
Although the family N[K]CC and KCC reciprocally regulate Cl− uptake for N[K]CCs and extrusion for KCCs, the mechanism of coupled ion translocation is considered to be similar. In an alternating access model for NKCCs proposed by Haas, the carriers only move when they are either fully loaded or completely empty[30]. Following ions binding in a strictly ordered sequence, a conformation change occurs that causes the transporter to face the opposite direction and adopt a new form (inwards facing state during extracellular ion binding and vice versa) [30]. The ions are subsequently released in the same order they were bound. Once empty, the transporters return to the original form ready for a new cycle. Recent 3D models have been made that support Haas, however, due to the many differences between paralogs of CCC family members, inferring functional properties from data obtained for a different family member may not be desirable[29].
2.2. Functional Regulation of the Cation Chloride Cotransporter Family
It is generally accepted that regulation of CCC function is accomplished through phosphorylation/dephosphorylation events. Phosphorylation/dephosphorylation reciprocally regulate the N[K]CC and KCC via a network of serine-threonine kinases and phosphatases[31][32][33]. Phosphorylation activates the Na+ dependent subfamily and inactivates the K+ dependent subfamily and vice versa [34]. The master regulator of CCCs, With-No-K (Lysine) kinases (WNKs) regulate the CCCs via their downstream targets, Ste20/SPS1-related proline/alanine-rich kinase (SPAK) or the SPAK homolog oxidative stress-responsive kinase 1 (OSR1)[31][32][33]. Threonine and serine residues of the CCCs are phosphorylated as a result of this downstream activation. The number and position of phospho-sites vary between different family members as they are not evolutionarily conserved among isoforms and paralogs[29]. Phosphatases such as protein phosphatase 1 (PP1) counterbalance the action of the kinases[35].
Recent data by Lee et al. [36]challenge the current understanding of KCC regulation. Lee and team reported that phosphorylation of KCC2 specific residue, Ser940, by protein kinase C (PKC) has been shown to activate KCC2, an isoform exclusively expressed in the nervous system. However, the phosphorylation of highly conserved threonine residues in all KCCs is ubiquitously associated with inactivation [33][37][38][39]. Thus, the phosphorylation-mediated-activation of KCC2 may be an adaptive mechanism to assist with KCC2 functions in the nervous system[38][40][41]. Gene mutations in WNK1 and WNK4 result in over-activation of the WNK-SPAK/OSR1 pathway, therefore increased phosphorylation and activation of NCC occurs in the kidney, resulting in hypertension[42][43]. Recent studies highlight multiple other signaling factors and regulators of CCC activities that are implicated in cardiovascular disease [44][45][46][47][48]. More specifically, the functional regulation of CCCs in vascular smooth muscle cells and renal cells play a significant role in the pathogenesis of atherosclerosis and Mendelian and non-Mendelian forms of hypertension[49][50][51].
2.3 Role of NCC in Cardiovascular Disease
Previous studies have demonstrated that the thiazide-sensitive NCC in the distal convoluted tubule (DCT) plays an important role in blood pressure regulation and that loss of NCC function causes hypotension (Figure 1). Contrastingly, gain-of-function mutations that result in the overactivation of NCC cause Familial Hyperkalemic Hypertension (FHHt) or Gordon’s syndrome, a Mendelian form of hypertension. FHHt is caused by genetic mutations in genes encoding for the regulatory WNK-SPAK/OSR1 and its upstream regulator, an E3 ubiquitin ligase complex containing kelch-like 3 (KLHL3) and Cullin 3 (CUL3). Indeed, current research is focused on identifying novel targets within the WNK-SPAK/OSR1 pathway for use in FHHt and potentially non-Mendelian forms of hypertension. A large body of evidence has demonstrated that inhibition of various components of the WNK-SPAK/OSR1-NCC pathway is an effective drug target for reducing blood pressure[52]. However, the lack of specificity across WNK and SPAK isoforms is challenging for the drug discovery process[45]. A more detailed account of the CUL3/KLHL3-WNK-SPAK/OSR1 pathway as a target for hypertension can be found in the review by Ferdaus et al[45].
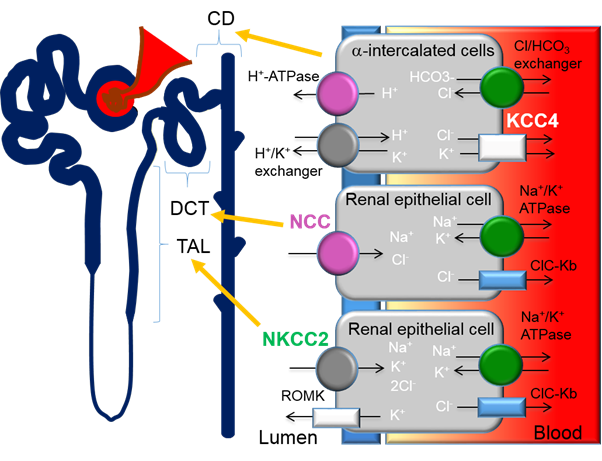
Figure 1. Expression of cation-chloride-cotransporters in the kidney. Alpha-intercalated cells in the collecting duct (CD) secretes acid via the apical H-ATPase and H+/K+ exchanger and reabsorbs bicarbonate via the basolateral Cl/HCO3 exchanger. Efflux of Cl− through the potassium chloride co-transporter-4 (KCC4) is important to maintain the electrochemical gradient to facilitate the acid secretion activities of the alpha-intercalated cells. Loss of KCC4 leads to renal tubular acidosis, which, if left untreated, could lead to cardiac arrhythmias. Sodium-chloride-cotransporters (NCCs) are exclusively expressed in the distal convoluted tubule (DCT). Gain-of-function mutations in regulatory genes that lead to the over activation of NCC cause Familial Hyperkalemic Hypertension (FHHt). Sodium potassium chloride cotransporters-2 (NKCC2) is expressed in the thick ascending limb (TAL). Loss-of-function mutations of NKCC2 results in the salt-wasting phenotype of type 1 Bartter Syndrome.
References
- BHF. BHF Heart and Circulatory Diseases in UK Statistics-Jul 2020 Factsheet. Availabe online: www.bhf.org.uk.
- WHO. A global brief on hypertension: Silent killer, global public health crisis: World Health Day 2013. World Health Organization (WHO). Availabe online: https://apps.who.int/iris/handle/10665/79059. (accessed on 7 October 2020).
- NHS. Cardiovascular disease (CVD) is a general term for conditions affecting the heart or blood vessels 2020. National Health Service (NHS). Availabe online: https://www.nhs.uk/conditions/cardiovascular-disease/. (accessed on 7 October 2020).
- Meor Azlan, N.; Koeners, M.; Zhang, J. Regulatory control of the Na-Cl co-transporter NCC and its therapeutic potential for hypertension. Acta Pharm. Sin. B. 2020, in press, doi:10.1016/j.apsb.2020.09.009.
- Schwartz, S.M.; Campbell, G.R.; Campbell, J.H. Replication of smooth muscle cells in vascular disease. Circ. Res. 1986, 58, 427–444, doi:10.1161/01.res.58.4.427.
- Zhang, J.; Bhuiyan, M.I.H.; Zhang, T.; Karimy, J.K.; Wu, Z.; Fiesler, V.M.; Zhang, J.; Huang, H.; Hasan, M.N.; Skrzypiec, A.E.; et al. Modulation of brain cation-Cl(-) cotransport via the SPAK kinase inhibitor ZT-1a. Nat. Commun 2020, 11, 78, doi:10.1038/s41467-019-13851-6.
- Hartmann, A.M.; Tesch, D.; Nothwang, H.G.; Bininda-Emonds, O.R. Evolution of the cation chloride cotransporter family: Ancient origins, gene losses, and subfunctionalization through duplication. Mol. Biol. Evol. 2014, 31, 434–447, doi:10.1093/molbev/mst225.
- Caron, L.; Rousseau, F.; Gagnon, E.; Isenring, P. Cloning and functional characterization of a cation-Cl- cotransporter-interacting protein. J. Biol Chem. 2000, 275, 32027–32036, doi:10.1074/jbc.M000108200.
- Wenz, M.; Hartmann, A.M.; Friauf, E.; Nothwang, H.G. CIP1 is an activator of the K+-Cl- cotransporter KCC2. Biochem. Biophys Res. Commun 2009, 381, 388–392, doi:10.1016/j.bbrc.2009.02.057.
- Hebert, S.C.; Mount, D.B.; Gamba, G. Molecular physiology of cation-coupled Cl- cotransport: The SLC12 family. Pflugers Arch. 2004, 447, 580–593, doi:10.1007/s00424-003-1066-3.
- Orlov, S.N.; Koltsova, S.V.; Kapilevich, L.V.; Gusakova, S.V.; Dulin, N.O. NKCC1 and NKCC2: The pathogenetic role of cation-chloride cotransporters in hypertension. Genes Dis. 2015, 2, 186–196, doi:10.1016/j.gendis.2015.02.007.
- Gamba, G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005, 85, 423–493, doi:10.1152/physrev.00011.2004.
- Markadieu, N.; Delpire, E. Physiology and pathophysiology of SLC12A1/2 transporters. Pflugers Arch. 2014, 466, 91–105, doi:10.1007/s00424-013-1370-5.
- Marshall-Phelps, K.L.H.; Kegel, L.; Baraban, M.; Ruhwedel, T.; Almeida, R.G.; Rubio-Brotons, M.; Klingseisen, A.; Benito-Kwiecinski, S.K.; Early, J.J.; Bin, J.M.; et al. Neuronal activity disrupts myelinated axon integrity in the absence of NKCC1b. J. Cell Biol. 2020, 219, e201909022, doi:10.1083/jcb.201909022.
- Merner, N.D.; Mercado, A.; Khanna, A.R.; Hodgkinson, A.; Bruat, V.; Awadalla, P.; Gamba, G.; Rouleau, G.A.; Kahle, K.T. Gain-of-function missense variant in SLC12A2, encoding the bumetanide-sensitive NKCC1 cotransporter, identified in human schizophrenia. J. Psychiatr Res. 2016, 77, 22–26, doi:10.1016/j.jpsychires.2016.02.016.
- Savardi, A.; Borgogno, M.; Narducci, R.; La Sala, G.; Ortega, J.A.; Summa, M.; Armirotti, A.; Bertorelli, R.; Contestabile, A.; De Vivo, M.; et al. Discovery of a Small Molecule Drug Candidate for Selective NKCC1 Inhibition in Brain Disorders. Chem 2020, 6, 2073–2096, doi:10.1016/j.chempr.2020.06.017.
- Plata, C.; Mount, D.B.; Rubio, V.; Hebert, S.C.; Gamba, G. Isoforms of the Na-K-2Cl cotransporter in murine TAL II. Functional characterization and activation by cAMP. Am. J. Physiol 1999, 276, F359–366, doi:10.1152/ajprenal.1999.276.3.F359.
- Vargas-Poussou, R.; Feldmann, D.; Vollmer, M.; Konrad, M.; Kelly, L.; van den Heuvel, L.P.; Tebourbi, L.; Brandis, M.; Karolyi, L.; Hebert, S.C.; et al. Novel molecular variants of the Na-K-2Cl cotransporter gene are responsible for antenatal Bartter syndrome. Am. J. Hum. Genet. 1998, 62, 1332–1340, doi:10.1086/301872.
- Simon, D.B.; Nelson-Williams, C.; Bia, M.J.; Ellison, D.; Karet, F.E.; Molina, A.M.; Vaara, I.; Iwata, F.; Cushner, H.M.; Koolen, M.; et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 1996, 12, 24–30, doi:10.1038/ng0196-24.
- Hunter, R.; Craigie, E.; Homer, N.; Mullins, J.; Bailey, M. Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am. J. Physiol Renal. Physiol. 2014, 306, F457–F467, doi:10.1152/ajprenal.00339.2013.
- Moreno, E.; Cristobal, P.S.; Rivera, M.; Vazquez, N.; Bobadilla, N.A.; Gamba, G. Affinity-defining domains in the Na-Cl cotransporter: A different location for Cl- and thiazide binding. J. Biol. Chem. 2006, 281, 17266–17275, doi:10.1074/jbc.M602614200.
- Gillen, C.M.; Brill, S.; Payne, J.A.; Forbush, B., 3rd. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J. Biol. Chem. 1996, 271, 16237–16244, doi:10.1074/jbc.271.27.16237.
- Kahle, K.T.; Merner, N.D.; Friedel, P.; Silayeva, L.; Liang, B.; Khanna, A.; Shang, Y.; Lachance-Touchette, P.; Bourassa, C.; Levert, A.; et al. Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 2014, 15, 766–774, doi:10.15252/embr.201438840.
- Delpire, E.; Baranczak, A.; Waterson, A.G.; Kim, K.; Kett, N.; Morrison, R.D.; Daniels, J.S.; Weaver, C.D.; Lindsley, C.W. Further optimization of the K-Cl cotransporter KCC2 antagonist ML077: Development of a highly selective and more potent in vitro probe. Bioorg Med. Chem. Lett. 2012, 22, 4532–4535, doi:10.1016/j.bmcl.2012.05.126.
- Pearson, M.M.; Lu, J.; Mount, D.B.; Delpire, E. Localization of the K(+)-Cl(-) cotransporter, KCC3, in the central and peripheral nervous systems: Expression in the choroid plexus, large neurons and white matter tracts. Neuroscience 2001, 103, 481–491, doi:10.1016/s0306-4522(00)00567-4.
- Howard, H.C.; Mount, D.B.; Rochefort, D.; Byun, N.; Dupre, N.; Lu, J.; Fan, X.; Song, L.; Riviere, J.B.; Prevost, C.; et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat. Genet. 2002, 32, 384–392, doi:10.1038/ng1002.
- Boettger, T.; Hubner, C.A.; Maier, H.; Rust, M.B.; Beck, F.X.; Jentsch, T.J. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 2002, 416, 874–878, doi:10.1038/416874a.
- Hewett, D.; Samuelsson, L.; Polding, J.; Enlund, F.; Smart, D.; Cantone, K.; See, C.G.; Chadha, S.; Inerot, A.; Enerback, C.; et al. Identification of a psoriasis susceptibility candidate gene by linkage disequilibrium mapping with a localized single nucleotide polymorphism map. Genomics 2002, 79, 305–314, doi:10.1006/geno.2002.6720.
- Hartmann, A.M.; Nothwang, H.G. Molecular and evolutionary insights into the structural organization of cation chloride cotransporters. Front. Cell Neurosci. 2014, 8, 470, doi:10.3389/fncel.2014.00470.
- Lytle, C.; McManus, T.J.; Haas, M. A model of Na-K-2Cl cotransport based on ordered ion binding and glide symmetry. Am. J. Physiol. 1998, 274, C299–309, doi:10.1152/ajpcell.1998.274.2.C299.
- Moriguchi, T.; Urushiyama, S.; Hisamoto, N.; Iemura, S.; Uchida, S.; Natsume, T.; Matsumoto, K.; Shibuya, H. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J. Biol Chem. 2005, 280, 42685–42693, doi:10.1074/jbc.M510042200.
- Vitari, A.C.; Deak, M.; Morrice, N.A.; Alessi, D.R. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem. J. 2005, 391, 17–24, doi:10.1042/BJ20051180.
- de Los Heros, P.; Alessi, D.R.; Gourlay, R.; Campbell, D.G.; Deak, M.; Macartney, T.J.; Kahle, K.T.; Zhang, J. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl- co-transporters. Biochem. J. 2014, 458, 559–573, doi:10.1042/BJ20131478.
- Rinehart, J.; Maksimova, Y.D.; Tanis, J.E.; Stone, K.L.; Hodson, C.A.; Zhang, J.; Risinger, M.; Pan, W.; Wu, D.; Colangelo, C.M.; et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 2009, 138, 525–536, doi:10.1016/j.cell.2009.05.031.
- Yang, X.; Wang, Q.; Cao, E. Structure of the human cation-chloride cotransporter NKCC1 determined by single-particle electron cryo-microscopy. Nat. Commun. 2020, 11, 1016, doi:10.1038/s41467-020-14790-3.
- Lee, H.H.; Walker, J.A.; Williams, J.R.; Goodier, R.J.; Payne, J.A.; Moss, S.J. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 2007, 282, 29777–29784, doi:10.1074/jbc.M705053200.
- Zhang, J.; Cordshagen, A.; Medina, I.; Nothwang, H.G.; Wisniewski, J.R.; Winklhofer, M.; Hartmann, A.M. Staurosporine and NEM mainly impair WNK-SPAK/OSR1 mediated phosphorylation of KCC2 and NKCC1. PLoS ONE 2020, 15, e0232967, doi:10.1371/journal.pone.0232967.
- Watanabe, M.; Zhang, J.; Mansuri, M.S.; Duan, J.; Karimy, J.K.; Delpire, E.; Alper, S.L.; Lifton, R.P.; Fukuda, A.; Kahle, K.T. Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 2019, 12, doi:10.1126/scisignal.aaw9315.
- Zhang, J.; Gao, G.; Begum, G.; Wang, J.; Khanna, A.R.; Shmukler, B.E.; Daubner, G.M.; de Los Heros, P.; Davies, P.; Varghese, J.; et al. Functional kinomics establishes a critical node of volume-sensitive cation-Cl(-) cotransporter regulation in the mammalian brain. Sci. Rep. 2016, 6, 35986, doi:10.1038/srep35986.
- Friedel, P.; Kahle, K.T.; Zhang, J.; Hertz, N.; Pisella, L.I.; Buhler, E.; Schaller, F.; Duan, J.; Khanna, A.R.; Bishop, P.N.; et al. WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci. Signal. 2015, 8, ra65, doi:10.1126/scisignal.aaa0354.
- Heubl, M.; Zhang, J.; Pressey, J.C.; Al Awabdh, S.; Renner, M.; Gomez-Castro, F.; Moutkine, I.; Eugene, E.; Russeau, M.; Kahle, K.T.; et al. GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl(-)-sensitive WNK1 kinase. Nat. Commun. 2017, 8, 1776, doi:10.1038/s41467-017-01749-0.
- Wilson, F.H.; Disse-Nicodeme, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.M.; et al. Human hypertension caused by mutations in WNK kinases. Science 2001, 293, 1107–1112, doi:10.1126/science.1062844.
- Wilson, F.H.; Kahle, K.T.; Sabath, E.; Lalioti, M.D.; Rapson, A.K.; Hoover, R.S.; Hebert, S.C.; Gamba, G.; Lifton, R.P. Molecular pathogenesis of inherited hypertension with hyperkalemia: The Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc. Natl. Acad. Sci. U S A 2003, 100, 680–684, doi:10.1073/pnas.242735399.
- Choe, K.Y.; Han, S.Y.; Gaub, P.; Shell, B.; Voisin, D.L.; Knapp, B.A.; Barker, P.A.; Brown, C.H.; Cunningham, J.T.; Bourque, C.W. High salt intake increases blood pressure via BDNF-mediated downregulation of KCC2 and impaired baroreflex inhibition of vasopressin neurons. Neuron 2015, 85, 549–560, doi:10.1016/j.neuron.2014.12.048.
- Ferdaus, M.Z.; McCormick, J.A. The CUL3/KLHL3-WNK-SPAK/OSR1 pathway as a target for antihypertensive therapy. Am. J. Physiol Renal Physiol 2016, 310, F1389–1396, doi:10.1152/ajprenal.00132.2016.
- Meyer, J.W.; Flagella, M.; Sutliff, R.L.; Lorenz, J.N.; Nieman, M.L.; Weber, C.S.; Paul, R.J.; Shull, G.E. Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na(+)-K(+)-2Cl(-) cotransporter. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1846–1855, doi:10.1152/ajpheart.00083.2002.
- Pace, A.J.; Lee, E.; Athirakul, K.; Coffman, T.M.; O’Brien, D.A.; Koller, B.H. Failure of spermatogenesis in mouse lines deficient in the Na(+)-K(+)-2Cl(-) cotransporter. J. Clin. Invest. 2000, 105, 441–450, doi:10.1172/JCI8553.
- Wang, Y.; Thatcher, S.; Cassis, L. Blood pressure monitoring using radio telemetry method in mice. Methods Mol. Biol. 2017, 1614, 75–85, doi:10.1007/978-1-4939-7030-8_7.
- O’Donnell, M.E.; Owen, N.E. Reduced Na-K-Cl cotransport in vascular smooth muscle cells from spontaneously hypertensive rats. Am. J. Physiol. 1988, 255, C169–180, doi:10.1152/ajpcell.1988.255.2.C169.
- Davis, J.P.; Chipperfield, A.R.; Harper, A.A. Accumulation of intracellular chloride by (Na-K-Cl) co-transport in rat arterial smooth muscle is enhanced in deoxycorticosterone acetate (DOCA)/salt hypertension. J. Mol. Cell Cardiol. 1993, 25, 233–237, doi:10.1006/jmcc.1993.1029.
- Jiang, G.; Akar, F.; Cobbs, S.L.; Lomashvilli, K.; Lakkis, R.; Gordon, F.J.; Sutliff, R.L.; O’Neill, W.C. Blood pressure regulates the activity and function of the Na-K-2Cl cotransporter in vascular smooth muscle. Am. J. Physiol. Heart. Circ. Physiol. 2004, 286, H1552–1557, doi:10.1152/ajpheart.00695.2003.
- Casu, G.; Merella, P. Diuretic Therapy in Heart Failure-Current Approaches. Eur. Cardiol. 2015, 10, 42–47, doi:10.15420/ecr.2015.10.01.42.