Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Melissa García Caballero and Version 2 by Catherine Yang.
The dynamic crosstalk between the different components of the tumor microenvironment is critical to determine cancer progression, metastatic dissemination, tumor immunity, and therapeutic responses. Angiogenesis is critical for tumor growth, and abnormal blood vessels contribute to hypoxia and acidosis in the tumor microenvironment. In this hostile environment, cancer and stromal cells have the ability to alter their metabolism in order to support the high energetic demands and favor rapid tumor proliferation.
- tumor microenvironment
- tumor angiogenesis
- tumor endothelial cell metabolism
1. Tumor Angiogenesis: A Pivotal Driver of Cancer Progression
Although solid tumor initiation does not rely on the formation of new blood vessels from the existing vascular bed (named angiogenesis), once the tumor is bigger than a few millimeters, it requires the formation of new blood vessels to ensure the supply of oxygen and nutrients, as well as to evacuate metabolic waste and carbon dioxide [1][2]. For several decades, sustained angiogenesis has been considered one of the hallmarks of cancer, as an excessive amount of proangiogenic stimuli in the TME maintains the “angiogenic switch” [2][22]. However, sprouting angiogenesis (SA) within tumors often results in nonproductive angiogenesis with abnormal blood vessels in structure and function [3][23]. In contrast to physiological conditions, in which the vasculature of the tissue efficiently supports blood distribution and transport from arteries to arterioles, and thereafter to capillaries, postcapillary venules, and veins, in tumors, there is a chaotic and nonhierarchical vasculature [4]. It is composed of abnormally dilated, tortuous, hyperpermeable, and hypoperfused vessels that lack proper perivascular coverage and tight EC junctions [5][24]. Thus, the presence of not fully functional vessels stimulates even more the production of proangiogenic factors (i.e., vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), placental growth factor (PlGF), angiopoietins, among others), consequently triggering a continuous self-reinforcing loop of nonproductive angiogenesis (reviewed by Cantelmo et al. [4] and Eelen et al. [3][23]).
The growing tumor creates high interstitial fluid pressure that collapses the blood vessels and induces fluid extravasation from the leaky tumor vessels [6][25]. This defective vasculature and the dysfunctional drainage, together with the hypoxia, low intratumoral pH and nutrient deprivation typical of the hostile TME, increase tumor aggressiveness and favor the escape of cancer cells [7][8][26,27]. Therefore, the tumor vasculature is not only required for the supply of nutrients and oxygen but also provides conduits for the dissemination of cancer cells from primary sites to other organs, giving rise to distant metastases. In addition, tumor vessels create a niche for cancer stem cells [9][10][28,29]. Besides cancer cells, cytokines and other factors in the TME, such as VEGF, can intravasate into the circulation and favor extravasation of metastatic cancer cells out of the blood vessels [11][30]. Immune cell infiltration in tumors is also determined by complex molecular and cellular mechanisms that limit or favor their penetration into the TME [12][31].
The formation of new blood vessels within the TME can be accomplished through different mechanisms [13][32]. The most common is SA, in which blood vessels grow toward a gradient of proangiogenic factors, such as VEGF-A [14][33]. This stimulus induces the transition of ECs from a quiescent to an activated state through the acquisition of a more migratory and invasive phenotype. Consequently, the release of metalloproteinases (MMPs) triggers the detachment of pericytes and the degradation of the basement membrane and ECM, allowing ECs to proliferate, migrate, and initiate a new sprout. ECs present in the newly formed vessel experience dynamic phenotypic adaptations to form tip, stalk, and phalanx cells. Tip cells are polarized cells extending lamellipodia and filopodia specialized in sensing their environment and guiding the new sprout, while stalk cells are more proliferative cells elongating the nascent vessel (Figure 1) [15][34]. Once the tip cells belonging to different sprouts meet, they anastomose, develop lumens that are surrounded by a basement membrane and form a perfused vessel, after which ECs differentiate into quiescent phalanx cells. Phalanx cells establish a barrier to ensure blood perfusion and maintain redox homeostasis. In parallel, mesenchymal cells differentiate into smooth muscle cells and pericytes that participate in the subsequent stabilization of the new blood vessel [16][35]. Of note, recent findings from the analysis of ECs at the single-cell level demonstrated a higher degree of phenotypic complexity and heterogeneity, including activated postcapillary venules, immature (characterized by the lack of specific marker gene expression, but expressing activation markers and upregulating ribosomal gene expression consistent with an activated intermediate phenotype), transitioning (refers to an intermediate immature EC to tip EC), breach cells (those ECs that create a breach in the basal lamina, presumably involved in the initiation of vessel sprouting by tip cells), and neophalanx (characterized by the expression of markers of mature capillaries and arteries and the upregulation of a Notch signaling gene signature) EC phenotypes, all of them associated with vessel formation under pathological situations [17][18][36,37].
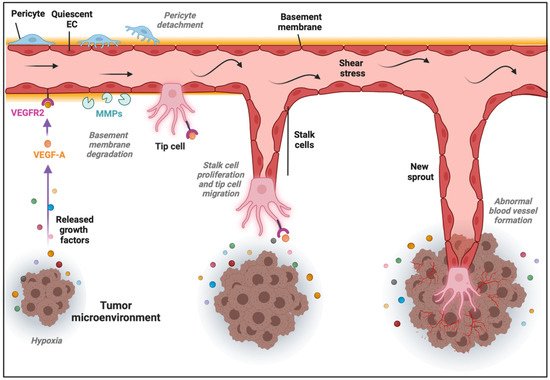
Figure 1. Sprouting angiogenesis in the tumor microenvironment (TME). Angiogenic stimuli such as hypoxia and proangiogenic growth factor gradients (in part produced by cancer cells) induce tip and stalk cell formation in a preexisting blood vessel. VEGF binds and activates its receptor VEGFR2, ECs become activated, and the detachment of pericytes and degradation of basement membrane and extracellular matrix (ECM) by matrix metalloproteinases (MMPs) take place. The tip cell becomes motile and starts to form lamellipodia and filopodia to migrate, while stalk cells proliferate to elongate the nascent vessel sprout in the TME.
Besides SA, there is an alternative mechanism of tumor angiogenesis, named intussusceptive angiogenesis (IA) [19][38]. IA is characterized by the longitudinal splitting of one existing vessel to originate new ones in order to expand the capillary plexus. During the IA process, two opposite ECs make kissing contact to form a transluminal bridge and interendothelial junctions are reorganized to finally form an interstitial pillar core. The latter is finally invaded by pericytes, myofibroblasts and mesenchymal cells, splitting the original vessel into two independent ones [19][38]. From a metabolic point of view, IA might be less demanding than SA, considering the low EC proliferation and migration, together with the faster pace with which ECM and perivascular cells promote rapid generation of new vessels. Although the exact molecular mechanisms of IA remain largely unknown, Notch signaling inhibition in the existing vascular bed promotes pericyte detachment and extravasation of mononuclear cells, leading to rapid vascular expansion by IA, while disruption of Notch signaling in the leading edges of the nascent vessel triggers SA [20][39]. Other possible mechanisms of IA have been reviewed elsewhere [21][22][23][40,41,42].
Additional nonangiogenic mechanisms of tumor vascularization can take place [24][43]. One of them is vessel co-option, in which cancer cells hijack preexisting quiescent vessels from the surrounding parenchymal tissue to be incorporated into the tumor mass [25][26][44,45]. ECs in co-opted vessels proliferate less than in angiogenic ones and show low expression levels of the typical angiogenic markers [25][44]. Vessel co-option, associated with a poor prognosis in patients, has been observed in primary and metastatic tumors, especially in lung, brain, and liver tumors, and is a resistance mechanism to antiangiogenic therapies [27][46]. Notably, single-cell RNA sequencing of > 30,000 cells from a lung-vessel co-option tumor model revealed that co-opted tumor ECs (TECs) and pericytes display a similar transcriptome signature to their normal counterparts [26][45]. Moreover, matrix-remodeling macrophages might help cancer cells to co-opt vessels, and an M1-like macrophage subtype might be involved in the maintenance of quiescent ECs (QECs) [26][45]. In another nonangiogenic process, cancer cells can behave as ECs and generate a vessel-like meshwork, a mechanism named vascular mimicry [28][47]. Although first observed in human uveal melanoma, vascular mimicry appears in different aggressive metastatic cancers, such as prostate, breast, and lung cancers, glioblastoma, and melanomas [29][30][31][48,49,50]. It has been postulated that hypoxia can induce this mechanism via epithelial-to-mesenchymal transition, thus allowing the generation of channels lined by cancer cells and embedded in a rich ECM that ensure the tumor blood supply and their connection with the surrounding vascular network [32][51].
2. Modulation of Tumor Endothelial Cell Metabolism as an Innovative Therapeutic Approach
A mechanism by which tumors can secure their blood supply is SA [33][34][35][53,109,110]. Years ago, this discovery led to the development of antiangiogenic treatments with the underlying idea that cutting off the blood supply would starve cancer cells to death. In some cancer types, this treatment works, but in most cases resistance eventually develops. Notably, VEGF or VEGF receptor blockade suffers from intrinsic refractoriness, acquired resistance, and inadequate efficacy [36][111]. Therefore, instead of targeting angiogenic signals, targeting EC metabolism to treat pathological angiogenesis has emerged as a new approach (Figure 24a), and several key metabolic enzymes, such as PFKFB3, CPT1a, asparagine synthetase (ASNS), GLS1, GS, FASN, PHGDH, OXCT1, and mitochondrial complex III enzymes, have been already investigated as potential targets to inhibit pathological (lymph)angiogenesis without causing systemic toxicity.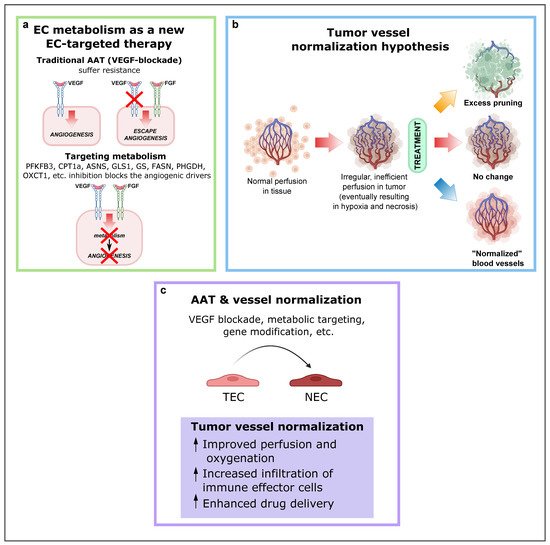
Figure 24. EC metabolism as a new EC-targeted therapy approach, tumor vessel normalization and alternative improved AAT in combination with other antitumor drugs. (a) Targeting EC metabolism as an alternative method to block pathological angiogenesis. While current anti-VEGF therapy compensatorily induces other angiogenic drivers (alternative growth factors), targeting EC metabolism, an engine driving angiogenesis, impairs vessel growth, regardless of how many angiogenic signals are present. Reproduced from [37][1] using BioRender.com. (b) Depending on the type and intensity of antiangiogenic therapy (AAT), the tumor vasculature can be pruned, leading to decreased blood perfusion (top); not respond to therapy (center); or become “normalized,” resulting in increased blood perfusion (bottom). Reproduced from Sorensen et al. [38][120] using BioRender.com. (c) Consequences and effects of AAT and vessel normalization. Abbreviations: EC, endothelial cell; VEGF, vascular endothelial growth factor; PFKFB3, phosphofructokinase-2/fructose-2,6-bisphosphatase 3; CPT1A, carnitine palmitoyltransferase 1A; ASNS, asparagine synthetase; GLS1, glutaminase; GS, glutamine synthase; FASN, fatty acid synthase; PHGDH, phosphoglycerate dehydrogenase; OXCT1, 3-oxoacid CoA-transferase 1; AAT, antiangiogenic therapies; TEC, tumor endothelial cell.
TECs are more glycolytic than NECs, and they upregulate PFKFB3 expression. Thus, lowering PFKFB3-driven glycolysis in TECs normalizes the abnormal tumor vasculature [39][40][63,67]. Both pharmacological and genetic inhibition can be used to block PFKFB3. Partial pharmacological inhibition with 3PO (targets PFKFB3) resulted in an in vivo and in vitro reduction of 35–40% of glycolysis in ECs [41][96]. Besides 3PO, other (more aggressive) glycolysis blockers have been tested, such as 2-deoxy-d-glucose (2DG), which reduced glycolysis up to 80%, but also caused cell death and an unhealthy morphology, while 3PO, on the other hand, did not provoke cell death. Since it has been demonstrated that only partial blockade of PFKFB3 is beneficial, drug dynamics should always be considered and not only the metabolic target itself.
Pharmacological blockade of CPT1a with etomoxir or perhexiline has shown promising antiangiogenic and antilymphangiogenic effects [42][43][44][45][56,82,112,113]. In the ocular retinopathy model (ROP model) in mouse pups and in the corneal model of injury-induced lymphangiogenesis, etomoxir reduced proliferating ECs and pathological vascular growth [42][43][56,82].
Several studies explored glutamine involvement in EC migration, sprouting and proliferation [39][46][63,84]. In particular, in vitro and in vivo GLS1 genetic deletion or GLS1-specific inhibitor (CB-839) inhibited EC migration, sprouting, and proliferation [46][84]. In the tumor environment, which is characterized by hypoxia, ER stress, and amino acid or glucose deficiency, ECs rely on ASNS in order to produce asparagine. ECs synthesize asparagine by converting glutamine-derived nitrogen and aspartate [47][48][49][50][51][114,115,116,117,118]. Therefore, GLS1 and ASNS are interesting metabolic targets to be considered in the antiangiogenic strategies.
Treatment with the FASN blocker orlistat caused antiangiogenic effects already at lower doses than that needed to impair cancer cell growth, offering a therapeutic window to use FASN inhibitors as an alternative antiangiogenic agent in cancer patients [52][12]. In agreement, in mice lacking endothelial FASN (FASNΔvEC), angiogenesis was impaired, and treatment with a pharmacological FASN inhibitor reduced pathological ocular neovascularization by decreasing EC proliferation [52][12]. Moreover, FASN inhibitors have antilymphangiogenic effects, decreasing LEC viability, proliferation, and migration, thereby impairing cancer cell lymphatic metastasis [53][119].
ECs use serine for the generation of dTTP, dATP and dGTP. PHGDH inhibition also impairs dCTP production indirectly as a consequence of reduced mitochondrial respiration, and impairs glycine production as a result of serine depletion [54][14]. Glycine is then required for heme synthesis in ECs, and the latter is crucial for the proper functioning of several enzymes, including the complexes II, III, and IV of OXPHOS mitochondrial respiration. As a result, the activity of mitochondrial dihydroorotate dehydrogenase, an enzyme involved in a key step in the production of dCTP, is decreased. Endothelial PHGDH silencing triggered severe oxidative stress, mitochondriopathy and impaired EC proliferation and sprouting in vitro. Moreover, decreased angiogenesis and neonatal lethality was observed in PHGDHECKO pups [54][14].
Genetic loss of OXCT1 in LECs impairs lymphangiogenesis, while ketone-body supplementation induces lymphatic growth in both developmental and pathological conditions [55][83]. In a mouse model of surgical ablation of lymphatic vessels in the tail, recapitulating characteristics of acquired lymphedema in humans, the ketogenic diet improved lymphatic vessel growth and lymph drainage and reduced lymphedema, suggesting a novel therapeutic opportunity through dietary metabolite supplements [37][55][1,83]. These findings may warrant future exploration of OXCT1 for inhibition of tumor-associated lymphangiogenesis and prevention of lymphatic metastasis.