Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Szandor Simmons.
Atherosclerosis—a systemic inflammatory disease—is the number one cause of mortality and morbidity worldwide. As such, the prevention of disease progression is of global interest in order to reduce annual deaths at a significant scale. Atherosclerosis is characterized by plaque formation in the arteries, resulting in vascular events such as ischemic stroke or myocardial infarction. Sphingolipids—a lipid class named after the chimeric creature sphinx—are considered to play a critical and, metaphorically, equally chimeric regulatory role in atherogenesis.
- cardiovascular disease
- atherosclerosis
- sphingolipids
- ceramide
- sphingosine-1-phosphate
1. Dihydroceramide in Atherosclerosis Progression
1.1. Synthesis and Metabolism
The de novo synthesis of sphingolipid is initiated by a highly coordinated sequence of actions involving serine palmitoyltransferase, 3-keto-dihydrosphingosine reductase, and dihydroceramide synthase, which convert cytosolic serine and palmitoyl CoA molecules via sphinganine into DhCer (Figure 21).
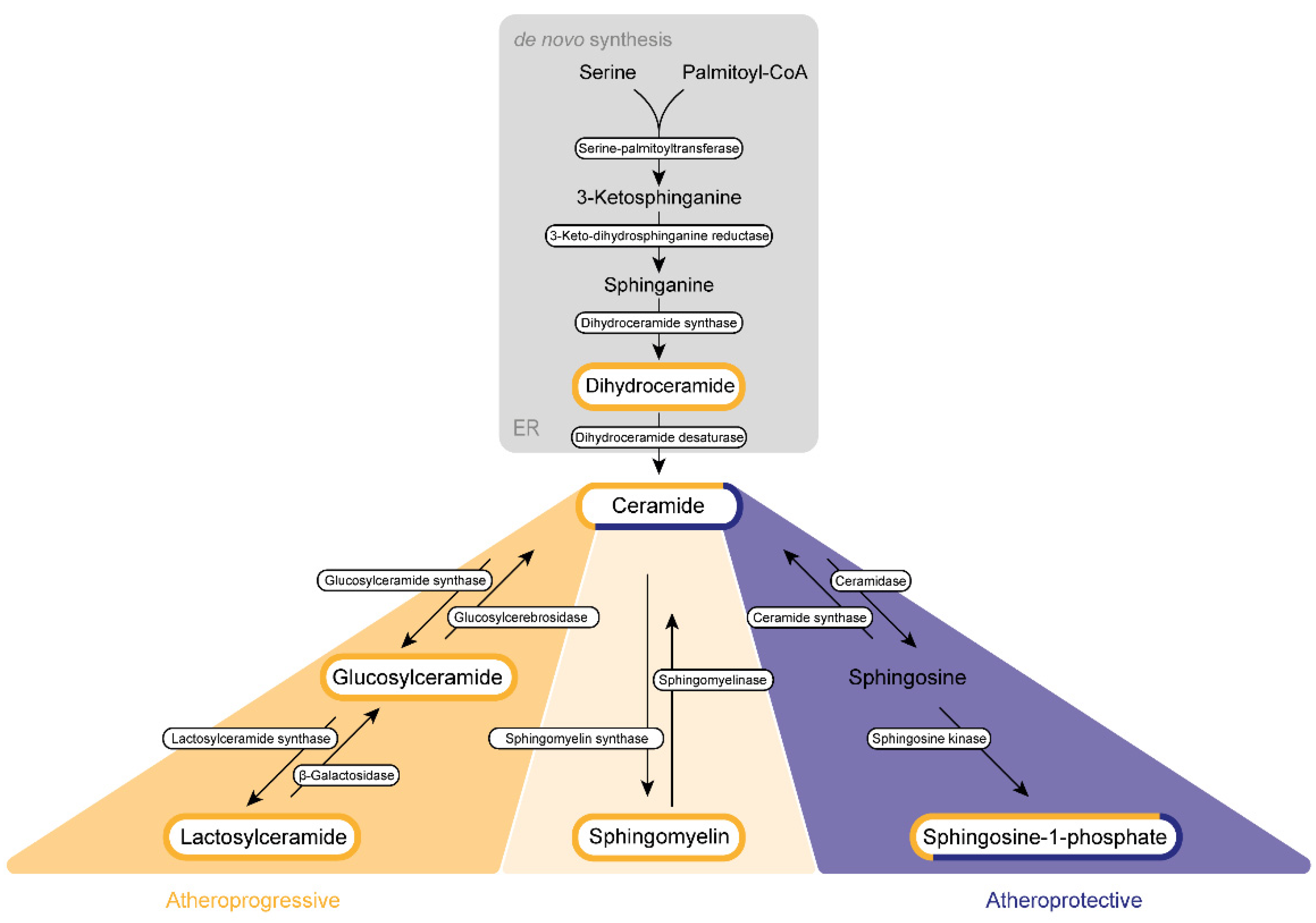
Figure 21. Sphingolipid biogenesis in atherosclerosis. Sphingolipids are synthesized de novo in the endoplasmic reticulum (ER) and the Golgi apparatus. Subsequently, they are transported via vesicles to the plasma membrane and the endosomes. The amino acid serine and palmitoyl-CoA provide the basis for the synthesis of 3-keto-sphinganine, which is reduced to sphinganine via 3-keto-dihydrosphinganine reductase. The dihydroceramide synthases form dihydroceramide, which can be catalyzed to ceramide, the backbone of all sphingolipids, by dihydroceramide desaturase. Ceramide itself can be converted into three further sphingolipid species. Glucosylceramide synthase mediates the production of glucosylceramide, which can be further modified to lactosylceramide through the enzyme lactosylceramide synthase. This modification can be reversed by β-galactosidase and glucosylcerebrosidase, respectively. Ceramide also provides the backbone for the generation of sphingomyelin via the activity of sphingomyelin synthase. Sphingosine-1-phosphate can be synthesized by ceramidase and sphingosine kinase. Several sphingolipids shown are assumed to exert influence on the progression of atherosclerosis. This impact can be categorized either as atherogenic (yellow) or as protective (purple) or can display characteristics of both categories (mixed).
DhCer is further processed at the endoplasmic reticulum (ER) membrane. Here, DhCer serves as a substrate for dihydroceramide desaturase that introduces a 4,5-trans-double bond to the sphingolipid backbone, thus generating Cer, which is further catalyzed by ceramidase and sphingosine kinases to first sphingosine and then S1P in the Golgi apparatus. Similar to most sphingolipids, DhCer is elevated in atherosclerotic plaques and is associated with inflammation and plaque instability [59][1].
1.2. Regulation of Inflammation
For a long time, no specific cellular function was attributed to DhCer, yet this notion has changed over the past 15 years, as DhCer was shown to impact autophagy, cell proliferation, cell survival and cell death in cancer and metabolic diseases [63,64,65,66,67][2][3][4][5][6]. In atheromatous plaques, DhCer levels positively correlate with proinflammatory cytokines such as monocyte chemoattractant protein-1, interleukin 6 (IL-6), and macrophage inflammatory protein-1 β. Over and above that, DhCer is able to induce the release of IL-6 in human coronary smooth muscle cells without inducing apoptosis [59][1]. However, caution is warranted in the interpretation of experimental results focusing on the specific function of DhCer, as pharmacological or genetic inhibition of enzymes involved in the de novo pathway will not only affect DhCer levels but also Cer concentration [68][7].
1.3. Regulation of Autophagy
In line with a potential functional role of DhCer in inflammatory processes per se, DhCer has been found to promote autophagy as demonstrated by the formation of autophagosomes in prostate cancer cells after stimulation with a DhCer desaturase inhibitor [69][8]. Of note, similar results were obtained by exogenous addition of short-chain DhCer [69][8]. Similarly, exogenous addition of DhCer analogues or treatment with DhCer desaturase inhibitors led to the accumulation of DhCer and promoted autophagy in cancer cells without causing cell death [64,65][3][4]. While a mechanistic link between DhCer and autophagy has thus been established, it remains a matter of controversy whether autophagy has a protective or a progressive effect on atherosclerosis. Normal autophagy flux is involved in vascular homeostasis, yet abnormal activity results in mechanisms aggravating atherosclerosis such as inducing thrombosis in endothelial cells, the secretion of pro-inflammatory cytokines by macrophages and abnormal remodeling of SMC in the intima. These characteristics can finally cause cell death and plaque instability [70][9]. Since short-chain DhCer can favor the formation of autophagosomes, it is appealing to hypothesize that short-chain DhCer also promotes autophagy in a pathophysiological context that may drive the progression of atherosclerosis. Moreover, the influence of DhCer on atherosclerosis promoting as well as atheroprotective mechanisms appears not to be restricted to autophagy only. DhCer has also been proposed to diminish apoptosis by inhibiting the formation of pores on the outer mitochondrial membrane, thereby impeding an essential step of the apoptotic cascade [71][10]. It remains to be evaluated whether and how this effect of DhCer on apoptosis influences atherosclerosis progression. In addition, DhCer affects oxidative stress by inducing ER stress. In contrast, DhCer levels are also elevated in the presence of oxidative stress, which can be explained by the inhibition of DhCer desaturase [72,73][11][12]. To investigate which effect provides the initiator for the other, further research is needed.
2. Sphingosine-1-Phosphate
The cleavage of fatty acids from the sphingolipid backbone of Cer by ceramidases releases sphingosine, which can be further phosphorylated by the activation of sphingosine kinase isoenzymes 1 and 2 (Sphk1, Sphk2) to spingosine-1-phosphate [131][13]. Sphk1 and Sphk2 are highly conserved and present in most mammalian cells and tissues, including platelets [132][14], erythrocytes [133][15], and the endothelium itself [134][16] which secrete S1P by the specific S1P-transporters major facilitator superfamily domain containing 2B (MFSD2B, erythrocytes and platelets) and spinster-homologue-2 (SPNS2, endothelial cells) into plasma and lymph [135,136,137,138][17][18][19][20]. Here, S1P signals as a bioactive lipid mediator by targeting five different G protein-coupled S1P-receptors (S1PR1-5) on various hematopoietic and vascular cells, and thereby controls cellular proliferation, apoptosis and cell migration in the blood vasculature and interstitial spaces and regulates endothelial barrier function [139][21]. Therefore, S1P/S1PR signaling may infer a significant role in the pathogenesis of atherosclerotic cardiovascular disease. Serum S1P is a strong and robust predictor of the occurrence of obstructive coronary artery disease [140][22], suggesting a correlation with atherogenic effects. Furthermore, the S1PR modulator FTY720, which acts upon all S1PRs except S1PR2 [141][23], effectively attenuates atherogenesis in ApoE- and LDL-receptor (LDL-R) deficient mice, respectively [142[24][25],143], implicating an atheroprotective effect. Future research should further confirm these contradictory initial findings. To realize signaling in health and disease, S1P has to bind to chaperone proteins including apolipoprotein M (ApoM) on HDL (~65% of all free plasma S1P) or albumin (~30% of all free plasma S1P) and LDL or VLDL (<5% of all plasma S1P), as its hydrophobic backbone and polar phosphate head group restrict the membrane permeability of S1P [144,145,146][26][27][28]. The plasma S1P levels also closely correlate to levels of total cholesterol, LDL cholesterol and HDL cholesterol in normolipidemic healthy subjects [147,148][29][30]. These associations may be of mutual functional relevance, e.g., the interaction of S1P and HDL has been proposed to reinforce their anti-thrombotic, anti-inflammatory and antioxidant properties [149][31]. The S1P/cholesterol interrelation has been experimentally validated by gain-of-function mutations of the LDL-R in livers of mice, which reduced S1P and ApoM levels in wildtype but not in ApoE-deficient mice. This finding suggests ApoE-dependent clearance of ApoM-associated S1P [150][32]. In line with this notion, statin treatment reduced serum ApoM levels in type 2 diabetes mellitus patients [151][33]. Further, only the S1P/ApoM complex on HDL is able to activate endothelial S1PR1 Gi-signaling and downstream ERK- and Akt-signaling, preserving endothelial adherent junctions [145][27] and decreasing TNFα-induced activation of nuclear factor kappa B (NFκB) and expression of ICAM-1 [152][34], while this endothelium-protective signaling cascade is only insufficiently activated by the S1P/albumin complex [153][35]. The majority of receptor-associated actions of S1PR-mediated intracellular processes are atheroprotective. Evidence from in vivo experiments shows that S1PR1 and S1PR3 are essential for both maintenance of endothelial barrier function, as the receptors’ downstream signaling cascade stabilizes endothelial cell–cell junctions [154][36] and attenuates endothelial contraction [155][37], and vascular relaxation by phosphorylation of eNOS and subsequently increased endothelial NO release [156,157,158][38][39][40]. In addition, a protective function against the development of atherosclerotic lesions has been suggested, as expression of the adhesion molecules VCAM-1 and ICAM-1 can be inhibited by S1PR1 signaling, thus reducing leukocyte adhesion and subsequent extravasation [157,158][39][40]. Analogously, S1P signaling via S1PR3 can inhibit the recruitment of inflammatory neutrophils and suppress apoptosis of cardiomyocytes. S1PR3-deficient mice are accordingly more susceptible for infarction in a mouse model of myocardial ischemia/reperfusion as compared to their corresponding wild type [159][41]. In contrast to this anti-inflammatory role of S1PR3 signaling, however, S1PR3 deficiency in ApoE-/- mice was found to strongly reduce monocyte recruitment by decreasing monocyte chemoattractant protein-1 secretion without affecting the size of atherogenic lesions [160][42]. These pro-inflammatory and, hence, potentially atherogenic properties of S1P signaling are further supported by the finding that S1PR1 enhances chemotaxis of lymphocytes and natural killer cells (NK) and, thus, has pro-inflammatory and pro-atherosclerotic properties [153][35]. S1P signaling through S1PR2 is even more likely to be associated with atherogenic functions. Although S1PR2 has been shown to inhibit SMC migration [161][43], it is centrally involved in the recruitment of inflammatory macrophages [162][44]. As such, S1PR2-/-/ApoE-/--double-deficient mice show reduced release of IL-18 and IL-1β, leading to impaired interstitial macrophage recruitment and, consequently, reduced formation of atherosclerotic plaques and necrotic cores in comparison to S1PR2-proficient mice [163][45]. Consistently, S1PR2-deficient macrophages express less CD36 and scavenger receptors ex vivo and increase cholesterol efflux while decreasing oxLDL uptake [163][45]. Atherogenic effects of S1PR2 signaling have also been suggested based on the fact that S1P can impair endothelial barrier function via the S1PR2/Rho/ROCK pathway [164][46]. However, S1PR2 deficiency in mice is associated with an increased risk of seizures and the development of B-cell lymphomas, arguing against the suitability of this receptor as a therapeutic target in atherosclerosis [165,166,167][47][48][49]. S1PR4, expressed on leukocytes, NK cells and airway SMC [168][50], and S1PR5 expressed on NK cells and oligodendrocytes [169][51] have not been associated with atheroprogression to date, even though S1PR4 stimulates IL-10 secretion from T-cells and simultaneously inhibits interleukin 4 and interferon-γ production [170][52], while S1PR5 mobilizes NK cells during infections. Although these findings may suggest an indirect involvement of S1PR4 and S1PR5 in atherosclerosis-associated inflammation, the latter receptors seem to have less of a direct impact on atherosclerosis pathology.3. Sphingomyelin (SM)
SM is the most abundant sphingolipid in mammalian tissues, where it serves as an important structural component of cell and plasma membranes [171][53]. Importantly, in the context of atherosclerosis, SM is also involved in maintaining cholesterol homoeostasis, as addition of exogenous SM to cells increases cholesterol biosynthesis and affects LDL binding to cell surface receptors [172,173][54][55]. However, there is further evidence implicating SM in the pathogenesis of atherosclerosis. SM has been identified as one component of human atherosclerotic plaques, and its abundance correlates with histological markers of plaque instability and is associated with the expression of pro-inflammatory cytokines. In accordance with this observation, stimulation of human coronary smooth muscle cells with SM in vitro induces a pro-inflammatory response reflected by IL-6 release [59][1]. SM plasma levels of atherosclerotic ApoE-/- mice are also elevated in comparison to WT mice [174][56]. Likewise, rabbits with hypercholesterolemia show elevated levels of SM compared with other lipids in atherosclerotic lesions [175][57]. Similar to S1P, SM in plasma is associated with VLDL/HDL cholesterol (63–75%) and LDL cholesterol (25–35%). The emerging notion that elevated SM levels in plasma are associated with pro-atherogenic properties is further supported by the fact that a decrease in HDL SM content is associated with smaller and more dense HDL. These complex lipoprotein particles favor cholesterol efflux, anti-oxidative activity toward LDL oxidation, antithrombotic activity in human platelets, as well as anti-inflammatory and anti-apoptotic activity [176][58]. In accordance, anti-apoptotic and anti-oxidative activities of small compact HDL cholesterol have been associated with SM degradation [177][59]. Unlike SMase, which hydrolyzes SM to Cer, the sphingomyelin synthase (SMS) catalyzes the synthesis of SM from Cer. SMS represents a family of different isoforms: SMS1 is primarily localized in the Golgi apparatus, whereas SMS2 primarily in plasma membranes [178,179][60][61]. Inhibition of SMS1 has been proposed as a potential therapeutic approach in atherosclerosis, as SMS1-/- mice show a decreased atherosclerotic phenotype characterized by reduced atherosclerotic lesions in the entire aortas as well as decreased macrophage content in these lesions [180][62]. Similar effects have been achieved in SMS2-deficient mice. These mice are marked by a reduction in secretion of pro-inflammatory cytokines, which is accompanied by the reduction of atherosclerotic lesions, necrotic core formation, macrophage content and collagen content compared to wild-type mice [181][63]. The pro-atherogenic capabilities of SM are further confirmed, as adenovirus-mediated insertion of SMS2 in ApoE -/- mice results in an increase in atherosclerotic lesions [182][64]. Similarly, SMS2 is shown to act as a modulator of NF-κB activation in HEK193 cells and macrophages from SMS2-deficent mice. This could provide one mechanistic explanation of the pro-atherogenic function of SM [183][65]. Consistent with this pro-atherogenic character of SM, overexpression of SMS1 and SMS2 increases the lipoprotein atherogenic potential in mice [184][66], whereas the simultaneous deficiency of SMS1 and SMS2 leads to a reduction in plasma SM and pro-inflammatory cytokine secretion [180][62]. In this context, it is remarkable that the inhibition of SMS1 alone leads to a decrease in the SM content in plasma, but simultaneously to an increase in DhCer and Cer in the plasma. Considering those two being associated with both atheroprotective and atherogenic effects, an explicit categorization of SM as an atheroprotective should only be made with caution. Further, it will be crucial to determine the mechanistic interplay between the inhibition of SMS1 and the increase in DhCer and Cer in order to identify a definite therapeutic signaling cascade. With regard to the identification of potential novel therapeutic targets, it is furthermore relevant to consider that loss-of-function by deletion of SMS1 (similar to S1PR2, vide supra) entails serious side effects such as low-frequency hearing loss [179[61][67],185], impaired insulin secretion [186][68], or CD4+ cell dysfunction [187][69].References
- Edsfeldt, A.; Dunér, P.; Ståhlman, M.; Mollet, I.G.; Asciutto, G.; Grufman, H.; Nitulescu, M.; Persson, A.F.; Fisher, R.M.; Melander, O. Sphingolipids contribute to human atherosclerotic plaque inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1132–1140.
- Zheng, W.; Kollmeyer, J.; Symolon, H.; Momin, A.; Munter, E.; Wang, E.; Kelly, S.; Allegood, J.C.; Liu, Y.; Peng, Q.; et al. Ceramides and other bioactive sphingolipid backbones in health and disease: Lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim. Biophys. Acta 2006, 1758, 1864–1884.
- Gagliostro, V.; Casas, J.; Caretti, A.; Abad, J.L.; Tagliavacca, L.; Ghidoni, R.; Fabrias, G.; Signorelli, P. Dihydroceramide delays cell cycle G1/S transition via activation of ER stress and induction of autophagy. Int. J. Biochem. Cell Biol. 2012, 44, 2135–2143.
- Signorelli, P.; Munoz-Olaya, J.M.; Gagliostro, V.; Casas, J.; Ghidoni, R.; Fabriàs, G. Dihydroceramide intracellular increase in response to resveratrol treatment mediates autophagy in gastric cancer cells. Cancer Lett. 2009, 282, 238–243.
- Venant, H.; Rahmaniyan, M.; Jones, E.E.; Lu, P.; Lilly, M.B.; Garrett-Mayer, E.; Drake, R.R.; Kraveka, J.M.; Smith, C.D.; Voelkel-Johnson, C. The Sphingosine Kinase 2 Inhibitor ABC294640 Reduces the Growth of Prostate Cancer Cells and Results in Accumulation of Dihydroceramides In Vitro and In Vivo. Mol. Cancer 2015, 14, 2744–2752.
- Breen, P.; Joseph, N.; Thompson, K.; Kraveka, J.M.; Gudz, T.I.; Li, L.; Rahmaniyan, M.; Bielawski, J.; Pierce, J.S.; van Buren, E.; et al. Dihydroceramide desaturase knockdown impacts sphingolipids and apoptosis after photodamage in human head and neck squamous carcinoma cells. Anticancer Res. 2013, 33, 77–84.
- Lachkar, F.; Ferré, P.; Foufelle, F.; Papaioannou, A. Dihydroceramides: Their emerging physiological roles and functions in cancer and metabolic diseases. Am. J. Physiol.-Endocrinol. Metab. 2021, 320, E122–E130.
- Jiang, Q.; Rao, X.; Kim, C.Y.; Freiser, H.; Zhang, Q.; Jiang, Z.; Li, G. Gamma-tocotrienol induces apoptosis and autophagy in prostate cancer cells by increasing intracellular dihydrosphingosine and dihydroceramide. Int. J. Cancer 2012, 130, 685–693.
- Hassanpour, M.; Rahbarghazi, R.; Nouri, M.; Aghamohammadzadeh, N.; Safaei, N.; Ahmadi, M. Role of autophagy in atherosclerosis: Foe or friend? J. Inflamm. 2019, 16, 1–10.
- Stiban, J.; Fistere, D.; Colombini, M. Dihydroceramide hinders ceramide channel formation: Implications on apoptosis. Apoptosis 2006, 11, 773–780.
- Magaye, R.R.; Savira, F.; Hua, Y.; Kelly, D.J.; Reid, C.; Flynn, B.; Liew, D.; Wang, B.H. The role of dihydrosphingolipids in disease. Cell. Mol. Life Sci. 2019, 76, 1107–1134.
- Siddique, M.M.; Li, Y.; Chaurasia, B.; Kaddai, V.A.; Summers, S.A. Dihydroceramides: From bit players to lead actors. J. Biol. Chem. 2015, 290, 15371–15379.
- Stunff, H.L.; Milstien, S.; Spiegel, S. Generation and metabolism of bioactive sphingosine-1-phosphate. J. Cell. Biochem. 2004, 92, 882–899.
- Yatomi, Y.; Ruan, F.; Hakomori, S.-I.; Igarashi, Y. Sphingosine-1-phosphate: A platelet-activating sphingolipid released from agonist-stimulated human platelets. Blood 1995, 86, 193–202.
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.-W.; Huang, Y.; Cyster, J.G. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298.
- Venkataraman, K.; Lee, Y.-M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 2008, 102, 669–676.
- Fukuhara, S.; Simmons, S.; Kawamura, S.; Inoue, A.; Orba, Y.; Tokudome, T.; Sunden, Y.; Arai, Y.; Moriwaki, K.; Ishida, J.; et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Investig. 2012, 122, 1416–1426.
- Simmons, S.; Sasaki, N.; Umemoto, E.; Uchida, Y.; Fukuhara, S.; Kitazawa, Y.; Okudaira, M.; Inoue, A.; Tohya, K.; Aoi, K.; et al. High-endothelial cell-derived S1P regulates dendritic cell localization and vascular integrity in the lymph node. Elife 2019, 8, e41239.
- Vu, T.M.; Ishizu, A.N.; Foo, J.C.; Toh, X.R.; Zhang, F.; Whee, D.M.; Torta, F.; Cazenave-Gassiot, A.; Matsumura, T.; Kim, S.; et al. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature 2017, 550, 524–528.
- Kobayashi, N.; Kawasaki-Nishi, S.; Otsuka, M.; Hisano, Y.; Yamaguchi, A.; Nishi, T. MFSD2B is a sphingosine 1-phosphate transporter in erythroid cells. Sci. Rep. 2018, 8, 4969.
- Yanagida, K.; Hla, T. Vascular and Immunobiology of the Circulatory Sphingosine 1-Phosphate Gradient. Annu. Rev. Physiol. 2017, 79, 67–91.
- Deutschman, D.H.; Carstens, J.S.; Klepper, R.L.; Smith, W.S.; Page, M.T.; Young, T.R.; Gleason, L.A.; Nakajima, N.; Sabbadini, R.A. Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate. Am. Heart J. 2003, 146, 62–68.
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897.
- Keul, P.; Sattler, K.; Levkau, B. HDL and its sphingosine-1-phosphate content in cardioprotection. Heart Fail. Rev. 2007, 12, 301–306.
- Nofer, J.R.; Bot, M.; Brodde, M.; Taylor, P.J.; Salm, P.; Brinkmann, V.; van Berkel, T.; Assmann, G.; Biessen, E.A. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007, 115, 501–508.
- Xu, N.; Dahlbäck, B. A novel human apolipoprotein (apoM). J. Biol. Chem. 1999, 274, 31286–31290.
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnström, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618.
- Aoki, S.; Yatomi, Y.; Ohta, M.; Osada, M.; Kazama, F.; Satoh, K.; Nakahara, K.; Ozaki, Y. Sphingosine 1-phosphate-related metabolism in the blood vessel. J. Biochem. 2005, 138, 47–55.
- Ohkawa, R.; Nakamura, K.; Okubo, S.; Hosogaya, S.; Ozaki, Y.; Tozuka, M.; Osima, N.; Yokota, H.; Ikeda, H.; Yatomi, Y. Plasma sphingosine-1-phosphate measurement in healthy subjects: Close correlation with red blood cell parameters. Ann. Clin. Biochem. 2008, 45, 356–363.
- Zhang, B.; Tomura, H.; Kuwabara, A.; Kimura, T.; Miura, S.; Noda, K.; Okajima, F.; Saku, K. Correlation of high density lipoprotein (HDL)-associated sphingosine 1-phosphate with serum levels of HDL-cholesterol and apolipoproteins. Atherosclerosis 2005, 178, 199–205.
- Levkau, B. HDL-S1P: Cardiovascular functions, disease-associated alterations, and therapeutic applications. Front. Pharmacol. 2015, 6, 243.
- Kurano, M.; Tsukamoto, K.; Hara, M.; Ohkawa, R.; Ikeda, H.; Yatomi, Y. LDL receptor and ApoE are involved in the clearance of ApoM-associated sphingosine 1-phosphate. J. Biol. Chem. 2015, 290, 2477–2488.
- Kappelle, P.J.; Ahnström, J.; Dikkeschei, B.D.; de Vries, R.; Sluiter, W.J.; Wolffenbuttel, B.H.; van Tol, A.; Nielsen, L.B.; Dahlbäck, B.; Dullaart, R.P. Plasma apolipoprotein M responses to statin and fibrate administration in type 2 diabetes mellitus. Atherosclerosis 2010, 213, 247–250.
- Galvani, S.; Sanson, M.; Blaho, V.A.; Swendeman, S.L.; Obinata, H.; Conger, H.; Dahlbäck, B.; Kono, M.; Proia, R.L.; Smith, J.D. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci. Signal. 2015, 8, ra79.
- Kurano, M.; Yatomi, Y. Sphingosine 1-phosphate and atherosclerosis. J. Atheroscler. Thromb. 2017, 25, 16–26.
- Argraves, K.M.; Gazzolo, P.J.; Groh, E.M.; Wilkerson, B.A.; Matsuura, B.S.; Twal, W.O.; Hammad, S.M.; Argraves, W.S. High density lipoprotein-associated sphingosine 1-phosphate promotes endothelial barrier function. J. Biol. Chem. 2008, 283, 25074–25081.
- Filep, J.G.; Földes-Filep, É.; Sirois, P. Nitric oxide modulates vascular permeability in the rat coronary circulation. Br. J. Pharmacol. 1993, 108, 323–326.
- Nofer, J.-R.; Van Der Giet, M.; Tölle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Gödecke, A.; Ishii, I.; Kleuser, B. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P 3. J. Clin. Investig. 2004, 113, 569–581.
- Kimura, T.; Tomura, H.; Mogi, C.; Kuwabara, A.; Damirin, A.; Ishizuka, T.; Sekiguchi, A.; Ishiwara, M.; Im, D.-S.; Sato, K. Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells. J. Biol. Chem. 2006, 281, 37457–37467.
- Ruiz, M.; Frej, C.; Holmér, A.; Guo, L.J.; Tran, S.; Dahlbäck, B. High-density lipoprotein–associated apolipoprotein M limits endothelial inflammation by delivering sphingosine-1-phosphate to the sphingosine-1-phosphate receptor 1. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 118–129.
- Theilmeier, G.; Schmidt, C.; Herrmann, J.R.; Keul, P.; Schäfers, M.; Herrgott, I.; Mersmann, J.; Larmann, J.; Hermann, S.; Stypmann, J.R. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation 2006, 114, 1403–1409.
- Keul, P.; Lucke, S.; von Wnuck Lipinski, K.; Bode, C.; Gräler, M.; Heusch, G.; Levkau, B. Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ. Res. 2011, 108, 314–323.
- Tamama, K.; Tomura, H.; Sato, K.; Malchinkhuu, E.; Damirin, A.; Kimura, T.; Kuwabara, A.; Murakami, M.; Okajima, F. High-density lipoprotein inhibits migration of vascular smooth muscle cells through its sphingosine 1-phosphate component. Atherosclerosis 2005, 178, 19–23.
- Duenas, A.I.; Aceves, M.; Fernández-Pisonero, I.; Gómez, C.; Orduna, A.; Crespo, M.S.; García-Rodríguez, C. Selective attenuation of Toll-like receptor 2 signalling may explain the atheroprotective effect of sphingosine 1-phosphate. Cardiovasc. Res. 2008, 79, 537–544.
- Skoura, A.; Michaud, J.; Im, D.-S.; Thangada, S.; Xiong, Y.; Smith, J.D.; Hla, T. Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 81–85.
- Sanchez, T.; Skoura, A.; Wu, M.T.; Casserly, B.; Harrington, E.O.; Hla, T. Induction of vascular permeability by the sphingosine-1-phosphate receptor–2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1312–1318.
- MacLennan, A.J.; Carney, P.R.; Zhu, W.J.; Chaves, A.H.; Garcia, J.; Grimes, J.R.; Anderson, K.J.; Roper, S.N.; Lee, N. An essential role for the H218/AGR16/Edg-5/LPB2 sphingosine 1-phosphate receptor in neuronal excitability. Eur. J. Neurosci. 2001, 14, 203–209.
- Cattoretti, G.; Mandelbaum, J.; Lee, N.; Chaves, A.H.; Mahler, A.M.; Chadburn, A.; Dalla-Favera, R.; Pasqualucci, L.; MacLennan, A.J. Targeted disruption of the S1P2 sphingosine 1-phosphate receptor gene leads to diffuse large B-cell lymphoma formation. Cancer Res. 2009, 69, 8686–8692.
- Green, J.A.; Suzuki, K.; Cho, B.; Willison, L.D.; Palmer, D.; Allen, C.D.; Schmidt, T.H.; Xu, Y.; Proia, R.L.; Coughlin, S.R. The sphingosine 1-phosphate receptor S1P 2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat. Immunol. 2011, 12, 672–680.
- Rivera, J.; Proia, R.L.; Olivera, A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat. Rev. Immunol. 2008, 8, 753–763.
- Terai, K.; Soga, T.; Takahashi, M.; Kamohara, M.; Ohno, K.; Yatsugi, S.; Okada, M.; Yamaguchi, T. Edg-8 receptors are preferentially expressed in oligodendrocyte lineage cells of the rat CNS. Neuroscience 2003, 116, 1053–1062.
- Wang, W.; Graeler, M.H.; Goetzl, E.J. Type 4 sphingosine 1-phosphate G protein-coupled receptor (S1P4) transduces S1P effects on T cell proliferation and cytokine secretion without signaling migration. FASEB J. 2005, 19, 1731–1733.
- Slotte, P.J. Molecular properties of various structurally defined sphingomyelins–correlation of structure with function. Prog. Lipid Res. 2013, 52, 206–219.
- Patton, S. Correlative relationship of cholesterol and sphingomyelin in cell membranes. J. Theor. Biol. 1970, 29, 489–491.
- Slotte, J.P. Biological functions of sphingomyelins. Prog. Lipid Res. 2013, 52, 424–437.
- Jeong, T.; Schissel, S.L.; Tabas, I.; Pownall, H.J.; Tall, A.R.; Jiang, X.-C. Increased sphingomyelin content of plasma lipoproteins in apolipoprotein E knockout mice reflects combined production and catabolic defects and enhances reactivity with mammalian sphingomyelinase. J. Clin. Investig. 1998, 101, 905–912.
- Bojic, L.A.; McLaren, D.G.; Shah, V.; Previs, S.F.; Johns, D.G.; Castro-Perez, J.M. Lipidome of atherosclerotic plaques from hypercholesterolemic rabbits. Int. J. Mol. Sci. 2014, 15, 23283–23293.
- Camont, L.; Lhomme, M.; Rached, F.; Le Goff, W.; Nègre-Salvayre, A.; Salvayre, R.; Calzada, C.; Lagarde, M.; Chapman, M.J.; Kontush, A. Small, dense high-density lipoprotein-3 particles are enriched in negatively charged phospholipids: Relevance to cellular cholesterol efflux, antioxidative, antithrombotic, anti-inflammatory, and antiapoptotic functionalities. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2715–2723.
- Mäkinen, V.-P.; Tynkkynen, T.; Soininen, P.; Forsblom, C.; Peltola, T.; Kangas, A.J.; Groop, P.-H.; Ala-Korpela, M. Sphingomyelin is associated with kidney disease in type 1 diabetes (The FinnDiane Study). Metabolomics 2012, 8, 369–375.
- Adachi, R.; Ogawa, K.; Matsumoto, S.-i.; Satou, T.; Tanaka, Y.; Sakamoto, J.; Nakahata, T.; Okamoto, R.; Kamaura, M.; Kawamoto, T. Discovery and characterization of selective human sphingomyelin synthase 2 inhibitors. Eur. J. Med. Chem. 2017, 136, 283–293.
- Yu, Z.; Peng, Q.; Huang, Y. Potential therapeutic targets for atherosclerosis in sphingolipid metabolism. Clin. Sci. 2019, 133, 763–776.
- Li, Z.; Fan, Y.; Liu, J.; Li, Y.; Huan, C.; Bui, H.H.; Kuo, M.S.; Park, T.S.; Cao, G.; Jiang, X.C. Impact of sphingomyelin synthase 1 deficiency on sphingolipid metabolism and atherosclerosis in mice. Arter. Thromb. Vasc. Biol. 2012, 32, 1577–1584.
- Liu, J.; Huan, C.; Chakraborty, M.; Zhang, H.; Lu, D.; Kuo, M.-S.; Cao, G.; Jiang, X.-C. Macrophage sphingomyelin synthase 2 deficiency decreases atherosclerosis in mice. Circ. Res. 2009, 105, 295–303.
- Wang, X.; Dong, J.; Zhao, Y.; Li, Y.; Wu, M. Adenovirus-mediated sphingomyelin synthase 2 increases atherosclerotic lesions in ApoE KO mice. Lipids Health Dis. 2011, 10, 1–5.
- Hailemariam, T.K.; Huan, C.; Liu, J.; Li, Z.; Roman, C.; Kalbfeisch, M.; Bui, H.H.; Peake, D.A.; Kuo, M.-S.; Cao, G. Sphingomyelin synthase 2 deficiency attenuates NFκB activation. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1519–1526.
- Dong, J.; Liu, J.; Lou, B.; Li, Z.; Ye, X.; Wu, M.; Jiang, X.-C. Adenovirus-mediated overexpression of sphingomyelin synthases 1 and 2 increases the atherogenic potential in mice. J. Lipid Res. 2006, 47, 1307–1314.
- Lu, M.H.; Takemoto, M.; Watanabe, K.; Luo, H.; Nishimura, M.; Yano, M.; Tomimoto, H.; Okazaki, T.; Oike, Y.; Song, W.J. Deficiency of sphingomyelin synthase-1 but not sphingomyelin synthase-2 causes hearing impairments in mice. J. Physiol. 2012, 590, 4029–4044.
- Yano, M.; Watanabe, K.; Yamamoto, T.; Ikeda, K.; Senokuchi, T.; Lu, M.; Kadomatsu, T.; Tsukano, H.; Ikawa, M.; Okabe, M. Mitochondrial dysfunction and increased reactive oxygen species impair insulin secretion in sphingomyelin synthase 1-null mice. J. Biol. Chem. 2011, 286, 3992–4002.
- Dong, L.; Watanabe, K.; Itoh, M.; Huan, C.-R.; Tong, X.-P.; Nakamura, T.; Miki, M.; Iwao, H.; Nakajima, A.; Sakai, T. CD4+ T-cell dysfunctions through the impaired lipid rafts ameliorate concanavalin A-induced hepatitis in sphingomyelin synthase 1-knockout mice. Int. Immunol. 2012, 24, 327–337.
More