Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Szandor Simmons and Version 2 by Catherine Yang.
Atherosclerosis—a systemic inflammatory disease—is the number one cause of mortality and morbidity worldwide. As such, the prevention of disease progression is of global interest in order to reduce annual deaths at a significant scale. Atherosclerosis is characterized by plaque formation in the arteries, resulting in vascular events such as ischemic stroke or myocardial infarction. Sphingolipids—a lipid class named after the chimeric creature sphinx—are considered to play a critical and, metaphorically, equally chimeric regulatory role in atherogenesis.
- cardiovascular disease
- atherosclerosis
- sphingolipids
- ceramide
- sphingosine-1-phosphate
1. Dihydroceramide in Atherosclerosis Progression
1.1. Synthesis and Metabolism
The de novo synthesis of sphingolipid is initiated by a highly coordinated sequence of actions involving serine palmitoyltransferase, 3-keto-dihydrosphingosine reductase, and dihydroceramide synthase, which convert cytosolic serine and palmitoyl CoA molecules via sphinganine into DhCer (Figure 12).
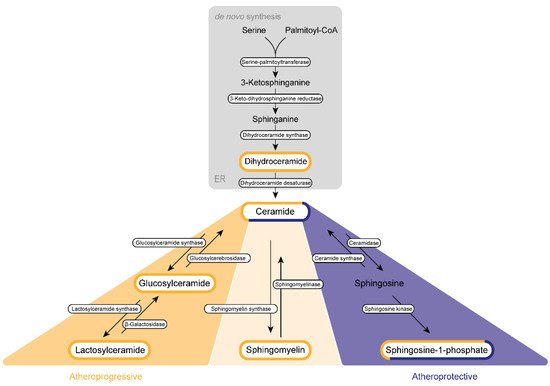
Figure 12. Sphingolipid biogenesis in atherosclerosis. Sphingolipids are synthesized de novo in the endoplasmic reticulum (ER) and the Golgi apparatus. Subsequently, they are transported via vesicles to the plasma membrane and the endosomes. The amino acid serine and palmitoyl-CoA provide the basis for the synthesis of 3-keto-sphinganine, which is reduced to sphinganine via 3-keto-dihydrosphinganine reductase. The dihydroceramide synthases form dihydroceramide, which can be catalyzed to ceramide, the backbone of all sphingolipids, by dihydroceramide desaturase. Ceramide itself can be converted into three further sphingolipid species. Glucosylceramide synthase mediates the production of glucosylceramide, which can be further modified to lactosylceramide through the enzyme lactosylceramide synthase. This modification can be reversed by β-galactosidase and glucosylcerebrosidase, respectively. Ceramide also provides the backbone for the generation of sphingomyelin via the activity of sphingomyelin synthase. Sphingosine-1-phosphate can be synthesized by ceramidase and sphingosine kinase. Several sphingolipids shown are assumed to exert influence on the progression of atherosclerosis. This impact can be categorized either as atherogenic (yellow) or as protective (purple) or can display characteristics of both categories (mixed).
DhCer is further processed at the endoplasmic reticulum (ER) membrane. Here, DhCer serves as a substrate for dihydroceramide desaturase that introduces a 4,5-trans-double bond to the sphingolipid backbone, thus generating Cer, which is further catalyzed by ceramidase and sphingosine kinases to first sphingosine and then S1P in the Golgi apparatus. Similar to most sphingolipids, DhCer is elevated in atherosclerotic plaques and is associated with inflammation and plaque instability [1][59].
1.2. Regulation of Inflammation
For a long time, no specific cellular function was attributed to DhCer, yet this notion has changed over the past 15 years, as DhCer was shown to impact autophagy, cell proliferation, cell survival and cell death in cancer and metabolic diseases [2][3][4][5][6][63,64,65,66,67]. In atheromatous plaques, DhCer levels positively correlate with proinflammatory cytokines such as monocyte chemoattractant protein-1, interleukin 6 (IL-6), and macrophage inflammatory protein-1 β. Over and above that, DhCer is able to induce the release of IL-6 in human coronary smooth muscle cells without inducing apoptosis [1][59]. However, caution is warranted in the interpretation of experimental results focusing on the specific function of DhCer, as pharmacological or genetic inhibition of enzymes involved in the de novo pathway will not only affect DhCer levels but also Cer concentration [7][68].
1.3. Regulation of Autophagy
In line with a potential functional role of DhCer in inflammatory processes per se, DhCer has been found to promote autophagy as demonstrated by the formation of autophagosomes in prostate cancer cells after stimulation with a DhCer desaturase inhibitor [8][69]. Of note, similar results were obtained by exogenous addition of short-chain DhCer [8][69]. Similarly, exogenous addition of DhCer analogues or treatment with DhCer desaturase inhibitors led to the accumulation of DhCer and promoted autophagy in cancer cells without causing cell death [3][4][64,65]. While a mechanistic link between DhCer and autophagy has thus been established, it remains a matter of controversy whether autophagy has a protective or a progressive effect on atherosclerosis. Normal autophagy flux is involved in vascular homeostasis, yet abnormal activity results in mechanisms aggravating atherosclerosis such as inducing thrombosis in endothelial cells, the secretion of pro-inflammatory cytokines by macrophages and abnormal remodeling of SMC in the intima. These characteristics can finally cause cell death and plaque instability [9][70]. Since short-chain DhCer can favor the formation of autophagosomes, it is appealing to hypothesize that short-chain DhCer also promotes autophagy in a pathophysiological context that may drive the progression of atherosclerosis. Moreover, the influence of DhCer on atherosclerosis promoting as well as atheroprotective mechanisms appears not to be restricted to autophagy only. DhCer has also been proposed to diminish apoptosis by inhibiting the formation of pores on the outer mitochondrial membrane, thereby impeding an essential step of the apoptotic cascade [10][71]. It remains to be evaluated whether and how this effect of DhCer on apoptosis influences atherosclerosis progression. In addition, DhCer affects oxidative stress by inducing ER stress. In contrast, DhCer levels are also elevated in the presence of oxidative stress, which can be explained by the inhibition of DhCer desaturase [11][12][72,73]. To investigate which effect provides the initiator for the other, further research is needed.