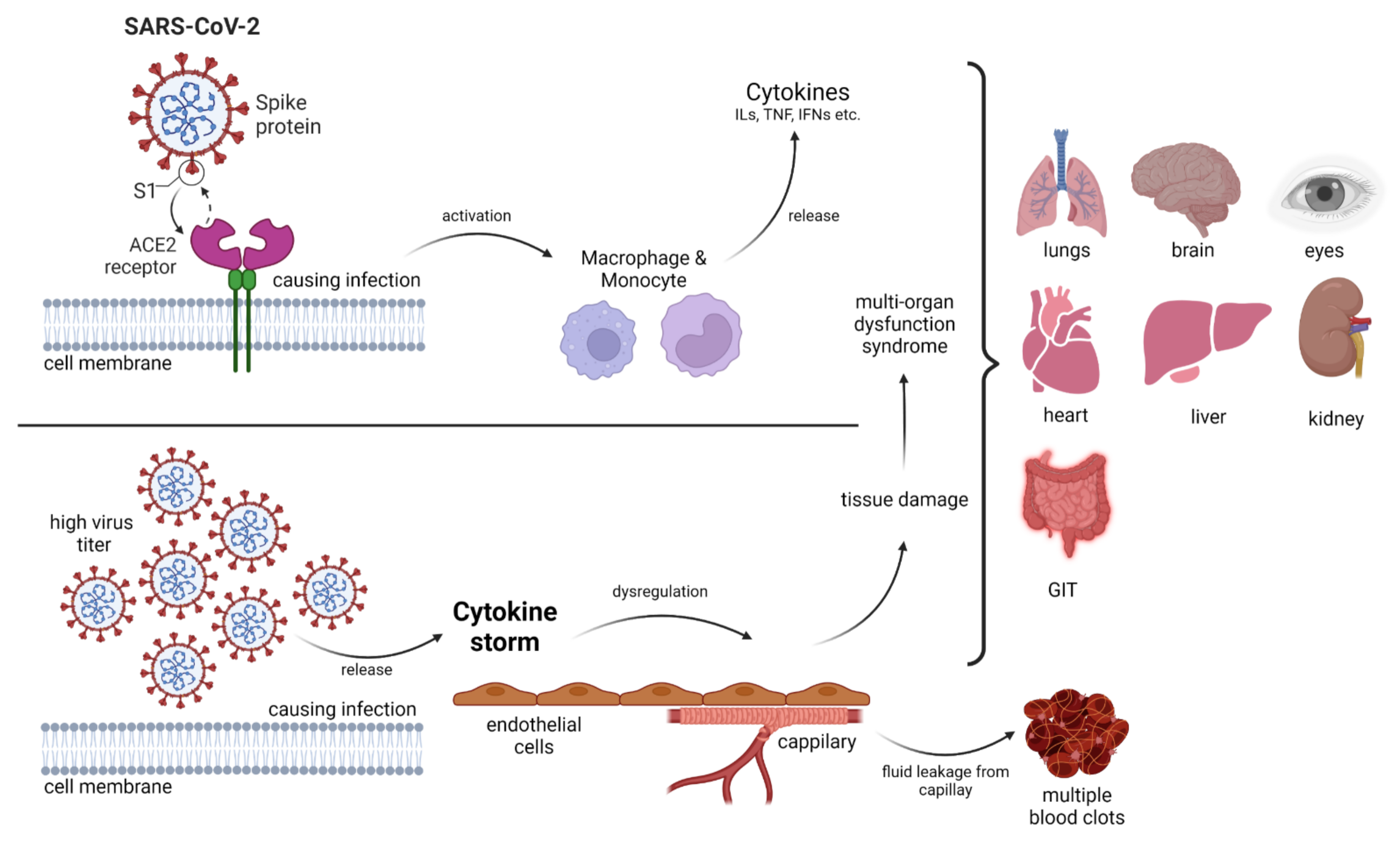
The innate immune system facilitates defense mechanisms against pathogen invasion and cell damage. Toll-like receptors (TLRs) assist in the activation of the innate immune system by binding to pathogenic ligands. This leads to the generation of intracellular signaling cascades including the biosynthesis of molecular mediators. TLRs on cell membranes are adept at recognizing viral components. Viruses can modulate the innate immune response with the help of proteins and RNAs that downregulate or upregulate the expression of various TLRs. In the case of COVID-19, molecular modulators such as type 1 interferons interfere with signaling pathways in the host cells, leading to an inflammatory response. Coronaviruses are responsible for an enhanced immune signature of inflammatory chemokines and cytokines. TLRs have been employed as therapeutic agents in viral infections as numerous antiviral Food and Drug Administration-approved drugs are TLR agonists.
- TLR
- immune system
- inflammation
- antiviral
- SARS-CoV-2
1. Introduction
2. Structure of Coronavirus
SARS-CoV-2, a member of the β-coronavirus genus in the family Coronaviridae, has an envelope and positive-sense ssRNA genome of 29,891 nucleotides, encoding circular nucleocapsid proteins with 9860 amino acid residues [21][18]. The viral particle size ranges from 80 to 220 nm. Overall, 10 open reading frames (ORFs) have been identified in its genome to date (approximately 26–32 kb). The first ORF (almost 2/3 of the viral RNA) encodes polyproteins 1a (ORF1a) and 1b (ORF1b) [22][19]. Furthermore, these ORFs are cleaved by proteases into 16 nonstructural proteins (NSPs) that are responsible for genome replication and transcription [23][20]. Structural proteins (SPs) are encoded by the remaining ORFs [24,25][21][22]. The main SPs and NSPs of SARS-CoV-2 are summarized in Table 1 and Table 2, respectively. The name coronavirus is derived from the appearance under the electron microscope, in which the presence of crown-like spikes on the envelope resembles the corona of the sun [26][23]. SPs form the viral envelope that holds the RNA genome, while NSPs are expressed in host-infected cells but are not incorporated into virion infectious particles. These NSPs include various transcription factors and enzymes such as RNA-dependent RNA polymerase (RdRp) and hemagglutinin esterase (HE). Moreover, the virion employs enzymes such as RNA replicases and viral proteases to replicate itself [22,27,28,29][19][24][25][26]. Various SPs have been identified including the glycoprotein membrane (M), spike (S), small envelope (E), and nucleoprotein (N), and other accessory proteins. M-glycoprotein is the most abundant, spanning the membrane bilayer thrice [30][27]. S-glycoprotein (150 kDa) is a type-I TM protein on the outer surface of the virus and is responsible for the binding of the virus to host cell receptors (ACE2). The S protein amino acid sequence of SARS-CoV-2 exhibits 86% similarity to that of SARS-CoV [31][28]. The S protein consists of oligosaccharides bound to serine amino acids through o-glycosides. The three major segments of S protein are the ectodomain, TM, and intracellular regions. The intracellular domain comprises the membrane fusion subunit S2 (trimeric stalk) as well as a short tail part known as the receptor-binding S1 domain (RBD; three S1 heads) [32,33][29][30]. Protein–protein interaction (PPI) between the human ACE2 and SARS-CoV-2 S protein facilitates viral attachment as well as the cellular entry of coronaviruses; thus, small-molecule blockage of these PPIs is a more inspiring therapeutic approach than inhibition via antibodies [34][31]. The S1 subunit of the S protein enables ACE2-mediated virus attachment, whereas the S2 subunit facilitates membrane fusion. Specifically, asparagine, glutamine, serine, phenylalanine, and leucine residues present in the S protein boost ACE2 binding [35][32]. Moreover, N protein bound to nucleic acids is an important structural component of the virus, which is responsible for viral replication and cellular response to infection in the host cellular machinery [31][28] (Table 1). The N protein comprises a serine-rich linker region sandwiched between the N-terminal domain (NTD) and the C-terminal domain (CTD). These termini are crucial for viral entry and processing in host cells. The CTD regulates nucleocapsid formation and the NTD adheres to the viral genome in the form of orthorhombic crystals. Phosphorylation sites are also present in the linker region, which control its function [35][32]. In the case of SARS-CoV, the N protein enhances the activation of cyclooxygenase-2 (COX-2), resulting in the inflammation of pulmonary cells [36][33]. Moreover, the N protein interacts with the p42 proteasome subunit, which degrades the virion [37][34]. This also disables type-I IFN, which is responsible for suppressing the host immune responses produced by biological systems against viral infections [38][35]. The interaction of the N protein with heterogeneous nuclear ribonucleoproteins leads to increased viral RNA synthesis [39][36]. The N protein sequence of SARS-CoV-2 shows a 94.3% similarity to that of the SARS-CoV [31][28]. The smallest TM structural protein in coronaviruses is the E protein (Table 1), which comprises two different domains: the NTD (1–9 residues) as well as a hydrophobic domain (10–37 residues), with a chain at the terminal (38–76 residues) [40,41,42][37][38][39]. The E protein plays a crucial biological role, not only in the structural integrity of the virus, but also in host virulence [43][40]. The E protein sequence of SARS-CoV-2 shows a 96.1% similarity to that of SARS-CoV [31][28]. The M protein plays a crucial role in maintaining the shape of the viral envelope (Table 1). This function can be achieved by interacting with other viral proteins that exhibit PPIs [44][41]. The M protein is also known as the central organization of coronavirus proteins. The binding of E to M produces the virus envelope, and this interaction is sufficient for the synthesis and release of viruses [45,46][42][43]. The binding of M with S is an important event for the retention of the S protein in the endoplasmic reticulum–Golgi complex as well as its integration into new viruses [46,47][43][44]. Moreover, the interaction of N with M stabilizes the nucleocapsid (RNA–N protein complex) and the internal core of viruses, resulting in the completion of viral assembly [47,48][44][45]. The M protein amino acid sequence of SARS-CoV-2 exhibits a 96.4% similarity with that of SARS-CoV [31][28].|
Sr. No. |
Sr. No. SPs |
PDB ID |
Residues |
NSPs Physiological Significance |
PDB ID |
Residues Reference |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Physiological Significance | Reference | ||||||||||
|
1 |
E |
7K3G |
|||||||||
|
1 |
NSP1 | 76–109 |
Virus assembly, morphogenesis, viral–host interaction, membrane permeability |
7K3N |
180 |
Protein synthesis, prevents antiviral activity of host cells, degrades host mRNA |
|||||
[ | , |
2 |
M |
8CTK |
220–260 |
638 Virus assembly, protein interactions (M–M, M–S, M–N) |
] |
||||
|
2 |
NSP2 |
7MSW Genome replication, disruption of intracellular host signaling |
3 |
N |
|||||||
|
3 | 6VY0, 6YUN |
NSP3 (Papain-like protease, PLpro) 422 |
7KAG, 6WEY, 6WUU, 7LG0 |
1945 Abundant RNA-binding protein, virion genome packaging |
Integral to viral replication, post-translational processing of the two polyproteins, suppresses host protein synthesis |
||||||
[ | , | [ |
4 | ||||
|
NSP4 |
3GZF |
500 |
Protects new replicated virions, replication and assembly of viral structures in host cell |
|
Drugs |
TLRs |
Viruses |
Significance |
References |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Pam2CSK4 |
TLR2 |
Parainfluenza |
Reduced virus replication |
- |
NF-κB activation and proinflammatory cytokines |
||||||||
|
TLR2 |
|||||||||||||
|
INNA-051 |
Hsp70, lipopeptide, HCV, Nonstructural protein 3 |
TLR2 Heterodimer |
SARS-CoV-2 Cell surface |
Reduces viral RNA load MyD88, Mal |
- |
NF-κB activation and proinflammatory cytokines |
|||||||
|
TLR3 |
dsRNA |
||||||||||||
|
PIKA |
TLR3 |
Homodimers |
Influenza A Endosomal membrane |
Reduces virus load TRIF |
[143][SARM negatively regulates TRIF |
IRF activation, production of type 1 IFNs and proinflammatory cytokines |
] |
||||||
|
TLR4 |
Lipopolysaccharide, Taxol, S protein of SARS-CoV-2 |
||||||||||||
|
Poly ICLC |
TLR3 | Homodimers |
HIV Cell surface |
Release of IFN-α/β/γ MyD88, Mal, TRIF, TRAM |
|||||||||
SARM negatively regulates TRIF and TRAM to consequently reduce inflammation | Activation of NF-κB, pro-inflammatory cytokines, and IFN-inducible genes | ] |
5 |
NSP5 (3C-like protease, 3CLpro) |
6LU7 |
306 |
|||||||
|
TLR5 | Protein cleavage capacity (conserved feature) |
Flagellin |
Homodimers |
Cell surface |
|||||||||
|
NA6 |
TLR4 |
Norovirus |
Induction of IFN-β |
MyD88 |
|||||||||
- | Activation of NF-κB and proinflammatory cytokines |
6 |
NSP6 |
- |
290 |
Induction of autophagosomes, inhibition of viral components to reach host lysosomes |
|||||||
|
TLR6 |
Diacyl lipopeptides, lipoteichoic acid, fungal zymosan | ] | [ | ][63] |
|||||||||
Heterodimer |
Cell surface |
MyD88, Mal/TIRAP |
- |
Activation of NF-κB and proinflammatory cytokines |
7 |
NSP7 |
7JLT |
83 |
Primase complex (NSP7-NSP8), hetero-oligomeric complex (NSP7-NSP8-RdRp), viral replication |
[67 | |||
|
TLR7 |
SARS-CoV-2 ssRNA, imadozoquinoline | , |
Homodimers |
Endosomal membrane , |
MyD88 |
||||||||
- | IRF activation, production of Type 1 IFNs and proinflammatory cytokines |
8 |
NSP8 |
||||||||||
|
TLR8 |
SARS-CoV-2 ssRNA |
7JLT |
198 |
Primase complex (NSP7-NSP8), hetero-oligomeric complex (NSP7-NSP8-RdRp), viral replication |
Endosomal membrane |
||||||||
MyD88 | - |
IRF activation, production of type 1 IFNs and proinflammatory cytokines |
9 |
NSP9 |
6WXD |
||||||||
|
TLR9 |
Unmethylated CPG-containing ssDNA, hemozoin from the malaria parasite | 113 |
Homodimers |
Endosomal membrane RNA synthesis, carries viral RNA to the host cell, responsible for proliferation |
MyD88 |
||||||||
- | IRF activation, production of type 1 IFNs and proinflammatory cytokines |
10 |
NSP10 |
6ZPE |
139 |
Cofactor activation for replicative enzymes, complex NSP10-NSP14, viral RNA proofreading |
|||||||
|
11 |
NSP11 |
- |
13 |
Cleavage product of PP1a by 3CLpro/MPro |
|||||||||
|
12 |
NSP12 (RNA polymerase, RdRp) |
6YYT |
932 |
RNA polymerase activity |
4. Role of Antiviral Drugs Employing TLRs
When a pathogen such as a virus invades, an antiviral immune response is evident in the host cells. Various conserved molecular patterns of PAMPs have been identified. As discussed above, TLRs are the key constituents of the innate immune system, and multiple TLRs (TLR1–4, TLR6–9) identify viral ligands [17,117,118,119][17][114][115][116]. With respect to their functional importance, TLRs might be potentially employed to treat not only inflammatory disorders but also viral diseases. This can be explained by a deep insight into the positive and negative mediators of TLRs [97,120][94][117]. TLR agonists lack accessory molecules but can mimic natural ligands; hence, they exhibit a low molecular weight and have potential for expanded pharmacokinetics and pharmacodynamics in comparison with the parent molecule. Moreover, TLR antagonists help to deal with autoimmune and inflammatory disorders by defeating unnecessary inflammation, resulting in an antibody- or cell-mediated response that suppresses disease progression [97,121,122][94][118][119]. Different approaches are employed by viruses in which they weaken their recognition by masking and/or increasing the dysregulation of mediators. Viruses disturb TLR signaling through their own mechanisms. Thus, TLRs are largely involved in the molecular interaction between viruses and host cells [5]. Various PRRs are engaged in the response to viral infection, which is also the case for TLRs. A thorough understanding of this interaction has facilitated the development of various strategies to limit viral infection including antiviral immunity as well as therapeutics [5]. Moreover, viral infection activates TLRs to increase cytokine levels, resulting in an antiviral innate immune response. The interaction between viruses and TLRs at every step of the signaling pathway plays an important role in developing effective antiviral therapies as well as in identifying novel molecular targets for the advancement in antiviral drugs [123][120]. The regulation of invasion, replication, and immune responses is a significant factor in viral pathogenesis [117][114]. Viral glycoproteins and NSPs released in the extracellular region are responsible for the stimulation of TLR2 and TLR4 due to their presence on the cellular surface [117,124,125][114][121][122]. In contrast, TLR3, TLR7/8, and TLR9, which are present in the endosomal compartment, contain viral double-stranded RNA (dsRNA) [126][123], ssRNA [114][111], and CpG DNA (unmethylated) [116][113], respectively. TLR agonists have a positive effect on antiviral immunity and exhibit significant resistance against experimental infections [127,128,129][124][125MPL | ||||||||||
TLR4 |
VZV |
Stimulate cytokines |
||||||||
|
Flagellin |
TLR5 |
Influenza A |
Reduces virus replication |
|||||||
|
CBLB502 |
TLR5 |
ConA |
Activation of NF-κB |
|||||||
|
Pam2CSK4 |
TLR6 |
Parainfluenza |
Reduces virus replication |
|||||||
|
INNA-051 |
TLR6 |
SARS-CoV-2 |
Reduces viral RNA load |
|||||||
|
GS-9620 |
TLR7 |
HIV |
Reactivates latency |
|||||||
[ | ||||||||||
|
Vesatolimod |
TLR7 |
HIV |
Modest delay in viral rebound |
][77] |
||||||
|
13 |
NSP13 |
|||||||||
|
R848 | 6JYT |
TLR7/8 601 |
Helicase activity |
[ |
Zika |
|||||
[ | ][147] |
14 |
NSP14 |
|||||||
|
GS-9688 | 7R2V |
527 |
Viral RNA methylation, viral RNA proofreading, methyltransferase activity |
[ |
TLR8 |
HBV |
Activation of dendritic and natural killer cells |
|||
|
15 |
NSP15 |
6WXC |
346 |
Endoribonuclease activity |
||||||
|
ODN2395 |
TLR9 |
Parainfluenza |
Reduces viral replication |
16 |
NSP16 |
6WVN |
298 |
Viral replication, immune response evasion Viral RNA methylation, methyltransferase activity |
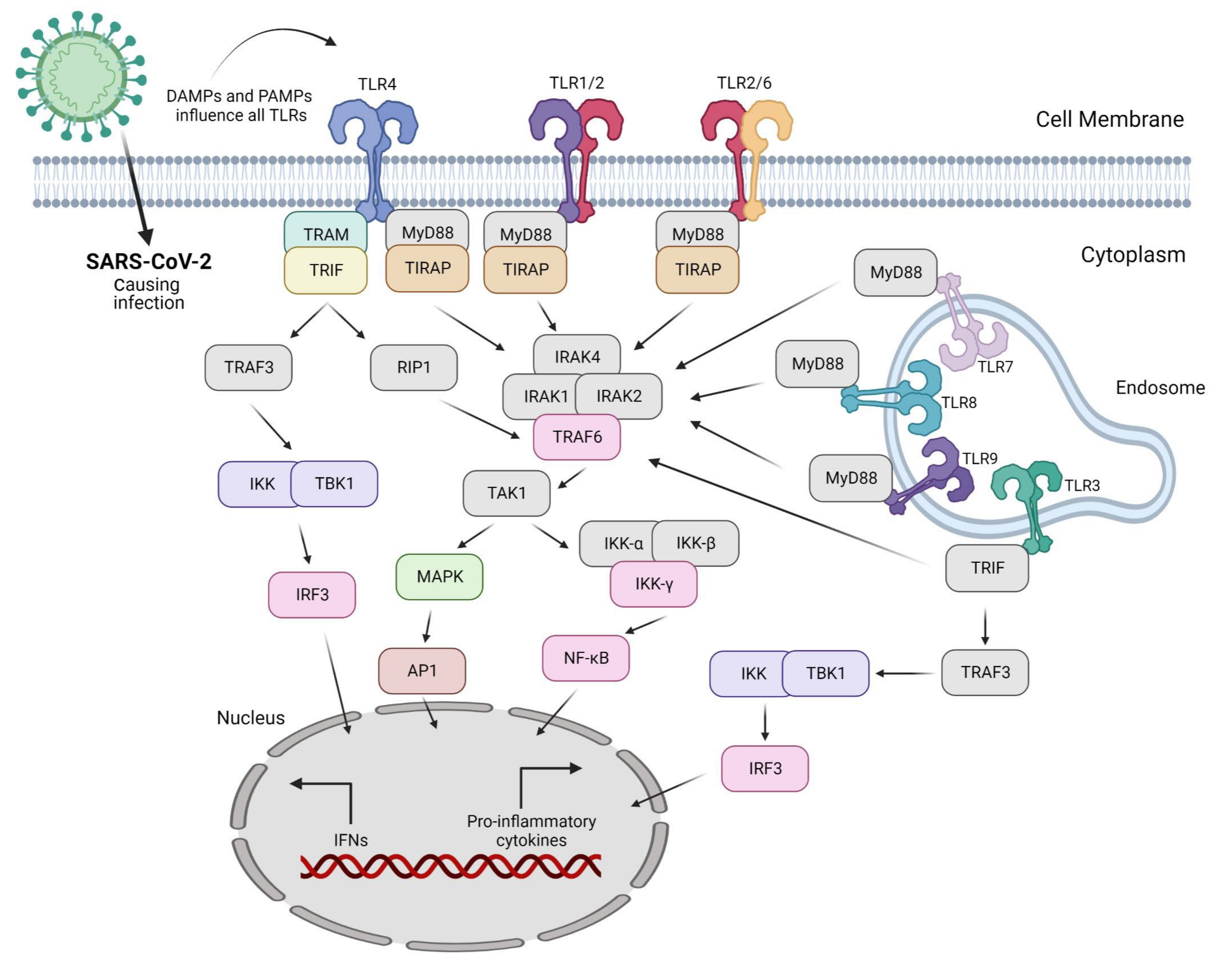
3. Overview of TLR Signaling
Invading pathogens stimulate the release of proinflammatory mediators in response to infection (Figure 1 and Figure 2). Signaling networks are necessary for the protection of the host against invading microorganisms. TLR signaling dysregulation plays a central role in the development and progression of infection. Inflammatory secretory molecules including chemokines, ILs, IFNs, and tumor necrosis factor-alpha (TNF-α) are part and parcel of TLR signaling, resulting in the modulation of cellular characteristics such as apoptosis, immune response, and proliferation [88,89,90][85][86][87]. Mitogen-activated protein kinases (MAPKs) and NF-κB are activated by TLRs. TLR3 and TLR4 are involved in the stimulation of IRF3. In contrast, IRF7 is triggered by TLR7–9 [91][88]. TLRs are stimulated by interactions with ligands to initiate an intracellular downstream signaling cascade, leading to activation of the host defense system [92CBLB502—Entolimod; ConA—Concanavalin A; GS-9688—Selgantolimod; R848—Resiquimod; NA6—neoagarohexaose; VZV—Varicella-Zoster virus.
References
- Iwasaki, A.; Medzhitov, R. Control of Adaptive Immunity by the Innate Immune System. Nat. Immunol. 2015, 16, 343–353.
- Patra, M.C.; Choi, S. Recent Progress in the Development of Toll-like Receptor (TLR) Antagonists. Expert Opin. Ther. Pat. 2016, 26, 719–730.
- Nie, L.; Cai, S.-Y.; Shao, J.-Z.; Chen, J. Toll-Like Receptors, Associated Biological Roles, and Signaling Networks in Non-Mammals. Front. Immunol. 2018, 9, 1523.
- Liu, G.; Zhang, H.; Zhao, C.; Zhang, H. Evolutionary History of the Toll-Like Receptor Gene Family across Vertebrates. Genome Biol. Evol. 2020, 12, 3615–3634.
- Carty, M.; Bowie, A.G. Recent Insights into the Role of Toll-like Receptors in Viral Infection. Clin. Exp. Immunol. 2010, 161, 397–406.
- Abe, T.; Kaname, Y.; Hamamoto, I.; Tsuda, Y.; Wen, X.; Taguwa, S.; Moriishi, K.; Takeuchi, O.; Kawai, T.; Kanto, T.; et al. Hepatitis C Virus Nonstructural Protein 5A Modulates the Toll-like Receptor-MyD88-Dependent Signaling Pathway in Macrophage Cell Lines. J. Virol. 2007, 81, 8953–8966.
- Li, S.Y.; Chen, C.; Zhang, H.Q.; Guo, H.Y.; Wang, H.; Wang, L.; Zhang, X.; Hua, S.N.; Yu, J.; Xiao, P.G.; et al. Identification of Natural Compounds with Antiviral Activities against SARS-Associated Coronavirus. Antivir. Res. 2005, 67, 18.
- Maloney, G.; Schröder, M.; Bowie, A.G. Vaccinia Virus Protein A52R Activates P38 Mitogen-Activated Protein Kinase and Potentiates Lipopolysaccharide-Induced Interleukin-10. J. Biol. Chem. 2005, 280, 30838–30844.
- Stack, J.; Haga, I.R.; Schröder, M.; Bartlett, N.W.; Maloney, G.; Reading, P.C.; Fitzgerald, K.A.; Smith, G.L.; Bowie, A.G. Vaccinia Virus Protein A46R Targets Multiple Toll-like-Interleukin-1 Receptor Adaptors and Contributes to Virulence. J. Exp. Med. 2005, 201, 1007–1018.
- Blasius, A.L.; Beutler, B. Intracellular Toll-like Receptors. Immunity 2010, 32, 305–315.
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733.
- Onofrio, L.; Caraglia, M.; Facchini, G.; Margherita, V.; de Placido, S.; Buonerba, C. Toll-like Receptors and COVID-19: A Two-Faced Story with an Exciting Ending. Future Sci. OA 2020, 6, FSO605.
- Pearce, L.; Davidson, S.M.; Yellon, D.M. The Cytokine Storm of COVID-19: A Spotlight on Prevention and Protection. Expert Opin. Ther. Targets 2020, 24, 723–730.
- Kaushik, D.; Bhandari, R.; Kuhad, A. TLR4 as a Therapeutic Target for Respiratory and Neurological Complications of SARS-CoV-2. Expert Opin. Ther. Targets 2021, 25, 491–508.
- Kawasaki, T.; Kawai, T. Toll-like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461.
- O’Neill, L.A.J.; Bowie, A.G. The Family of Five: TIR-Domain-Containing Adaptors in Toll-like Receptor Signalling. Nat. Rev. Immunol. 2007, 7, 353–364.
- Takeuchi, O.; Akira, S. Innate Immunity to Virus Infection. Immunol. Rev. 2009, 227, 75–86.
- Chan, J.F.W.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.W.; Yuan, S.; Yuen, K.Y. Genomic Characterization of the 2019 Novel Human-Pathogenic Coronavirus Isolated from a Patient with Atypical Pneumonia after Visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236.
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016, 96, 59.
- Shi, S.T.; Lai, M.M.C. Viral and Cellular Proteins Involved in Coronavirus Replication. In Coronavirus Replication and Reverse Genetics; Springer: Berlin/Heidelberg, Germany, 2005; Volume 287, p. 95.
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The Origin, Transmission and Clinical Therapies on Coronavirus Disease 2019 (COVID-19) Outbreak-an Update on the Status. Mil. Med. Res. 2020, 7, 11.
- Han, Y.; Du, J.; Su, H.; Zhang, J.; Zhu, G.; Zhang, S.; Wu, Z.; Jin, Q. Identification of Diverse Bat Alphacoronaviruses and Betacoronaviruses in China Provides New Insights into the Evolution and Origin of Coronavirus-Related Diseases. Front. Microbiol. 2019, 10, 1900.
- Park, S.E. Epidemiology, Virology, and Clinical Features of Severe Acute Respiratory Syndrome -Coronavirus-2 (SARS-CoV-2; Coronavirus Disease-19). Clin. Exp. Pediatr. 2020, 63, 119–124.
- Gasmalbari, E.; Abbadi, O.S. Non-Structural Proteins of SARS-CoV-2 as Potential Sources for Vaccine Synthesis. Infect. Dis. Trop. Med. 2020, 6, e667.
- Gorla, U.S.; Rao, G.S.N.K. SARS-CoV-2: The Prominent Role of Non-Structural Proteins (NSPS) in COVID-19. Indian J. Pharm. Educ. Res. 2020, 54, S381–S389.
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216.
- Mousavizadeh, L.; Ghasemi, S. Genotype and Phenotype of COVID-19: Their Roles in Pathogenesis. J. Microbiol. Immunol. Infect. 2021, 54, 159–163.
- Mohammed, M.E.A. The Percentages of SARS-CoV-2 Protein Similarity and Identity with SARS-CoV and BatCoV RaTG13 Proteins Can Be Used as Indicators of Virus Origin. J. Proteins Proteom. 2021, 12, 81.
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The Spike Protein of SARS-CoV--a Target for Vaccine and Therapeutic Development. Nat. Rev. Microbiol. 2009, 7, 226–236.
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM Structures of MERS-CoV and SARS-CoV Spike Glycoproteins Reveal the Dynamic Receptor Binding Domains. Nat. Commun. 2017, 8, 15092.
- Bojadzic, D.; Alcazar, O.; Chen, J.; Chuang, S.T.; Condor Capcha, J.M.; Shehadeh, L.A.; Buchwald, P. Small-Molecule Inhibitors of the Coronavirus Spike: ACE2 Protein-Protein Interaction as Blockers of Viral Attachment and Entry for SARS-CoV-2. ACS Infect. Dis. 2021, 7, 1519–1534.
- Satarker, S.; Nampoothiri, M. Structural Proteins in Severe Acute Respiratory Syndrome Coronavirus-2. Arch. Med. Res. 2020, 51, 482–491.
- Yan, X.; Hao, Q.; Mu, Y.; Timani, K.A.; Ye, L.; Zhu, Y.; Wu, J. Nucleocapsid Protein of SARS-CoV Activates the Expression of Cyclooxygenase-2 by Binding Directly to Regulatory Elements for Nuclear Factor-Kappa B and CCAAT/Enhancer Binding Protein. Int. J. Biochem. Cell Biol. 2006, 38, 1417–1428.
- Wang, Q.; Li, C.; Zhang, Q.; Wang, T.; Li, J.; Guan, W.; Yu, J.; Liang, M.; Li, D. Interactions of SARS Coronavirus Nucleocapsid Protein with the Host Cell Proteasome Subunit P42. Virol. J. 2010, 7, 99.
- Lu, X.; Pan, J.; Tao, J.; Guo, D. SARS-CoV Nucleocapsid Protein Antagonizes IFN-β Response by Targeting Initial Step of IFN-β Induction Pathway, and Its C-Terminal Region Is Critical for the Antagonism. Virus Genes 2011, 42, 37.
- Luo, H.; Chen, Q.; Chen, J.; Chen, K.; Shen, X.; Jiang, H. The Nucleocapsid Protein of SARS Coronavirus Has a High Binding Affinity to the Human Cellular Heterogeneous Nuclear Ribonucleoprotein A1. FEBS Lett. 2005, 579, 2623.
- Ruch, T.R.; Machamer, C.E. The Hydrophobic Domain of Infectious Bronchitis Virus E Protein Alters the Host Secretory Pathway and Is Important for Release of Infectious Virus. J. Virol. 2011, 85, 675–685.
- Kuo, L.; Hurst, K.R.; Masters, P.S. Exceptional Flexibility in the Sequence Requirements for Coronavirus Small Envelope Protein Function. J. Virol. 2007, 81, 2249–2262.
- Verdiá-Báguena, C.; Nieto-Torres, J.L.; Alcaraz, A.; Dediego, M.L.; Enjuanes, L.; Aguilella, V.M. Analysis of SARS-CoV E Protein Ion Channel Activity by Tuning the Protein and Lipid Charge. Biochim. Biophys. Acta 2013, 1828, 2026–2031.
- Venkatagopalan, P.; Daskalova, S.M.; Lopez, L.A.; Dolezal, K.A.; Hogue, B.G. Coronavirus Envelope (E) Protein Remains at the Site of Assembly. Virology 2015, 478, 75–85.
- Schoeman, D.; Fielding, B.C. Coronavirus Envelope Protein: Current Knowledge. Virol. J. 2019, 16, 69.
- Corse, E.; Machamer, C.E. Infectious Bronchitis Virus E Protein Is Targeted to the Golgi Complex and Directs Release of Virus-like Particles. J. Virol. 2000, 74, 4319–4326.
- Mortola, E.; Roy, P. Efficient Assembly and Release of SARS Coronavirus-like Particles by a Heterologous Expression System. FEBS Lett 2004, 576, 174–178.
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23.
- Narayanan, K.; Maeda, A.; Maeda, J.; Makino, S. Characterization of the Coronavirus M Protein and Nucleocapsid Interaction in Infected Cells. J. Virol. 2000, 74, 8127–8134.
- Liu, D.X.; Yuan, Q.; Liao, Y. Coronavirus Envelope Protein: A Small Membrane Protein with Multiple Functions. Cell Mol. Life Sci 2007, 64, 2043–2048.
- Arndt, A.L.; Larson, B.J.; Hogue, B.G. A Conserved Domain in the Coronavirus Membrane Protein Tail Is Important for Virus Assembly. J. Virol. 2010, 84, 11418–11428.
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 Nucleocapsid Protein Is Dynamic, Disordered, and Phase Separates with RNA. Nat. Commun. 2021, 12, 1936.
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S. wen Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149.
- Züst, R.; Cervantes-Barragán, L.; Kuri, T.; Blakqori, G.; Weber, F.; Ludewig, B.; Thiel, V. Coronavirus Non-Structural Protein 1 Is a Major Pathogenicity Factor: Implications for the Rational Design of Coronavirus Vaccines. PLoS Pathog. 2007, 3, 1062–1072.
- Afsar, M.; Narayan, R.; Akhtar, M.N.; Das, D.; Rahil, H.; Nagaraj, S.K.; Eswarappa, S.M.; Tripathi, S.; Hussain, T. Drug Targeting Nsp1-Ribosomal Complex Shows Antiviral Activity against SARS-CoV-2. Elife 2022, 11, e74877.
- Kamitani, W.; Narayanan, K.; Huang, C.; Lokugamage, K.; Ikegami, T.; Ito, N.; Kubo, H.; Makino, S. Severe Acute Respiratory Syndrome Coronavirus Nsp1 Protein Suppresses Host Gene Expression by Promoting Host MRNA Degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 12885–12890.
- Ma, J.; Chen, Y.; Wu, W.; Chen, Z. Structure and Function of N-Terminal Zinc Finger Domain of SARS-CoV-2 NSP2. Virol. Sin. 2021, 36, 1104–1112.
- Cornillez-Ty, C.T.; Liao, L.; Yates, J.R.; Kuhn, P.; Buchmeier, M.J. Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein 2 Interacts with a Host Protein Complex Involved in Mitochondrial Biogenesis and Intracellular Signaling. J. Virol. 2009, 83, 10314–10318.
- Angeletti, S.; Benvenuto, D.; Bianchi, M.; Giovanetti, M.; Pascarella, S.; Ciccozzi, M. COVID-2019: The Role of the Nsp2 and Nsp3 in Its Pathogenesis. J Med Virol 2020, 92, 584–588.
- Khan, M.T.; Zeb, M.T.; Ahsan, H.; Ahmed, A.; Ali, A.; Akhtar, K.; Malik, S.I.; Cui, Z.; Ali, S.; Khan, A.S.; et al. SARS-CoV-2 Nucleocapsid and Nsp3 Binding: An in Silico Study. Arch. Microbiol. 2021, 203, 59–66.
- Alsaadi, E.A.J.; Jones, I.M. Membrane Binding Proteins of Coronaviruses. Future Virol. 2019, 14, 275–286.
- Oostra, M.; te Lintelo, E.G.; Deijs, M.; Verheije, M.H.; Rottier, P.J.M.; de Haan, C.A.M. Localization and Membrane Topology of Coronavirus Nonstructural Protein 4: Involvement of the Early Secretory Pathway in Replication. J. Virol. 2007, 81, 12323–12336.
- Helmy, Y.A.; Fawzy, M.; Elaswad, A.; Sobieh, A.; Kenney, S.P.; Shehata, A.A. The COVID-19 Pandemic: A Comprehensive Review of Taxonomy, Genetics, Epidemiology, Diagnosis, Treatment, and Control. J. Clin. Med. 2020, 9, 1225.
- Stobart, C.C.; Sexton, N.R.; Munjal, H.; Lu, X.; Molland, K.L.; Tomar, S.; Mesecar, A.D.; Denison, M.R. Chimeric Exchange of Coronavirus Nsp5 Proteases (3CLpro) Identifies Common and Divergent Regulatory Determinants of Protease Activity. J. Virol. 2013, 87, 12611.
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary Analysis of SARS-CoV-2: How Mutation of Non-Structural Protein 6 (NSP6) Could Affect Viral Autophagy. J. Infect. 2020, 81, e24–e27.
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus NSP6 Restricts Autophagosome Expansion. Autophagy 2014, 10, 1426–1441.
- Sun, X.; Liu, Y.; Huang, Z.; Xu, W.; Hu, W.; Yi, L.; Liu, Z.; Chan, H.; Zeng, J.; Liu, X.; et al. SARS-CoV-2 Non-Structural Protein 6 Triggers NLRP3-Dependent Pyroptosis by Targeting ATP6AP1. Cell Death Differ. 2022, 29, 1240–1254.
- Biswal, M.; Diggs, S.; Xu, D.; Khudaverdyan, N.; Lu, J.; Fang, J.; Blaha, G.; Hai, R.; Song, J. Two Conserved Oligomer Interfaces of NSP7 and NSP8 Underpin the Dynamic Assembly of SARS-CoV-2 RdRP. Nucleic Acids Res. 2021, 49, 5956–5966.
- Krichel, B.; Falke, S.; Hilgenfeld, R.; Redecke, L.; Uetrecht, C. Processing of the SARS-CoV Pp1a/Ab Nsp7-10 Region. Biochem. J. 2020, 477, 1009–1019.
- Te Velthuis, A.J.W.; van den Worm, S.H.E.; Snijder, E.J. The SARS-Coronavirus Nsp7+nsp8 Complex Is a Unique Multimeric RNA Polymerase Capable of Both de Novo Initiation and Primer Extension. Nucleic Acids Res. 2012, 40, 1737–1747.
- Zeng, Z.; Deng, F.; Shi, K.; Ye, G.; Wang, G.; Fang, L.; Xiao, S.; Fu, Z.; Peng, G. Dimerization of Coronavirus Nsp9 with Diverse Modes Enhances Its Nucleic Acid Binding Affinity. J. Virol. 2018, 92, e00692-18.
- De Araújo, O.J.; Pinheiro, S.; Zamora, W.J.; Alves, C.N.; Lameira, J.; Lima, A.H. Structural, Energetic and Lipophilic Analysis of SARS-CoV-2 Non-Structural Protein 9 (NSP9). Sci. Rep. 2021, 11, 23003.
- Littler, D.R.; Gully, B.S.; Colson, R.N.; Rossjohn, J. Crystal Structure of the SARS-CoV-2 Non-Structural Protein 9, Nsp9. iScience 2020, 23, 101258.
- Rona, G.; Zeke, A.; Miwatani-Minter, B.; de Vries, M.; Kaur, R.; Schinlever, A.; Garcia, S.F.; Goldberg, H.V.; Wang, H.; Hinds, T.R.; et al. The NSP14/NSP10 RNA Repair Complex as a Pan-Coronavirus Therapeutic Target. Cell Death Differ. 2021, 29, 285–292.
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural Basis and Functional Analysis of the SARS Coronavirus Nsp14-Nsp10 Complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441.
- Bouvet, M.; Lugari, A.; Posthuma, C.C.; Zevenhoven, J.C.; Bernard, S.; Betzi, S.; Imbert, I.; Canard, B.; Guillemot, J.C.; Lécine, P.; et al. Coronavirus Nsp10, a Critical Co-Factor for Activation of Multiple Replicative Enzymes. J. Biol. Chem. 2014, 289, 25783–25796.
- Gadhave, K.; Kumar, P.; Kumar, A.; Bhardwaj, T.; Garg, N.; Giri, R. Conformational Dynamics of 13 Amino Acids Long NSP11 of SARS-CoV-2 under Membrane Mimetics and Different Solvent Conditions. Microb. Pathog. 2021, 158, 105041.
- Elfiky, A.A. SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) Targeting: An in Silico Perspective. J. Biomol. Struct. Dyn. 2021, 39, 3204–3212.
- Khan, M.T.; Irfan, M.; Ahsan, H.; Ahmed, A.; Kaushik, A.C.; Khan, A.S.; Chinnasamy, S.; Ali, A.; Wei, D.-Q. Structures of SARS-CoV-2 RNA-Binding Proteins and Therapeutic Targets. Intervirology 2021, 64, 55–68.
- Mishra, A.; Rathore, A.S. RNA Dependent RNA Polymerase (RdRp) as a Drug Target for SARS-CoV2. J. Biomol. Struct. Dyn. 2022, 40, 6039–6051.
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural Basis for Inhibition of the RNA-Dependent RNA Polymerase from SARS-CoV-2 by Remdesivir. Science 2020, 368, 1499.
- Wang, H.; Xue, S.; Yang, H.; Chen, C. Recent Progress in the Discovery of Inhibitors Targeting Coronavirus Proteases. Virol. Sin. 2016, 31, 24.
- Niu, X.; Kong, F.; Hou, Y.J.; Wang, Q. Crucial Mutation in the Exoribonuclease Domain of Nsp14 of PEDV Leads to High Genetic Instability during Viral Replication. Cell Biosci. 2021, 11, 106.
- Tahir, M. Coronavirus Genomic Nsp14-ExoN, Structure, Role, Mechanism, and Potential Application as a Drug Target. J. Med. Virol. 2021, 93, 4258–4264.
- Krafcikova, P.; Silhan, J.; Nencka, R.; Boura, E. Structural Analysis of the SARS-CoV-2 Methyltransferase Complex Involved in RNA Cap Creation Bound to Sinefungin. Nat. Commun. 2020, 11, 3717.
- Yoshimoto, F.K. A Biochemical Perspective of the Nonstructural Proteins (NSPs) and the Spike Protein of SARS CoV-2. Protein J. 2021, 40, 260–295.
- Vithani, N.; Ward, M.D.; Zimmerman, M.I.; Novak, B.; Borowsky, J.H.; Singh, S.; Bowman, G.R. SARS-CoV-2 Nsp16 Activation Mechanism and a Cryptic Pocket with Pan-Coronavirus Antiviral Potential. Biophys. J. 2021, 120, 2880–2889.
- Rosas-Lemus, M.; Minasov, G.; Shuvalova, L.; Inniss, N.L.; Kiryukhina, O.; Brunzelle, J.; Satchell, K.J.F. High-Resolution Structures of the SARS-CoV-2 2′-O-Methyltransferase Reveal Strategies for Structure-Based Inhibitor Design. Sci. Signal. 2020, 13, eabe1202.
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066.
- Balka, K.R.; de Nardo, D. Understanding Early TLR Signaling through the Myddosome. J. Leukoc. Biol. 2019, 105, 339–351.
- Satoh, T.; Akira, S. Toll-Like Receptor Signaling and Its Inducible Proteins. Microbiol. Spectr. 2016, 4, 4–6.
- Barton, G.M.; Kagan, J.C. A Cell Biological View of Toll-like Receptor Function: Regulation through Compartmentalization. Nat. Rev. Immunol. 2009, 9, 535–541.
- Wang, Y.; Song, E.; Bai, B.; Vanhoutte, P.M. Toll-like Receptors Mediating Vascular Malfunction: Lessons from Receptor Subtypes. Pharmacol. Ther. 2016, 158, 91–100.
- Kagan, J.C. Signaling Organelles of the Innate Immune System. Cell 2012, 151, 1168–1178.
- Yamamoto, M.; Sato, S.; Mori, K.; Hoshino, K.; Takeuchi, O.; Takeda, K.; Akira, S. Cutting Edge: A Novel Toll/IL-1 Receptor Domain-Containing Adapter That Preferentially Activates the IFN-Beta Promoter in the Toll-like Receptor Signaling. J. Immunol. 2002, 169, 6668–6672.
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 2010, 11, 373–384.
- Zhang, Y.; Liang, C. Innate Recognition of Microbial-Derived Signals in Immunity and Inflammation. Sci. China Life Sci. 2016, 59, 1210–1217.
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like Receptors Activation, Signaling, and Targeting: An Overview. Bull. Natl. Res. Cent. 2019, 43, 187.
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of Toll-like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front. Physiol. 2017, 8, 508.
- Lushpa, V.A.; Goncharuk, M.V.; Lin, C.; Zalevsky, A.O.; Talyzina, I.A.; Luginina, A.P.; Vakhrameev, D.D.; Shevtsov, M.B.; Goncharuk, S.A.; Arseniev, A.S.; et al. Modulation of Toll-like Receptor 1 Intracellular Domain Structure and Activity by Zn2+ Ions. Commun. Biol. 2021, 4, 1003.
- Takeuchi, O.; Sato, S.; Horiuchi, T.; Hoshino, K.; Takeda, K.; Dong, Z.; Modlin, R.L.; Akira, S. Cutting Edge: Role of Toll-Like Receptor 1 in Mediating Immune Response to Microbial Lipoproteins. J. Immunol. 2002, 169, 10–14.
- Ahmed, S.; Moawad, M.; Elhefny, R.; Chest, M.A.-E.J. Is Toll like Receptor 4 a Common Pathway Hypothesis for Development of Lung Cancer and Idiopathic Pulmonary Fibrosis? Elsevier: Amsterdam, The Netherlands, 2016.
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The Role of TLR2 Ininfection and Immunity. Front. Immunol. 2012, 3, 79.
- Zainol, M.I.B.; Kawasaki, T.; Monwan, W.; Murase, M.; Sueyoshi, T.; Kawai, T. Innate Immune Responses through Toll-like Receptor 3 Require Human-Antigen-R-Mediated Atp6v0d2 MRNA Stabilization. Sci. Rep. 2019, 9, 20406.
- Chen, Y.; Lin, J.; Zhao, Y.; Ma, X.; Yi, H. Toll-like Receptor 3 (TLR3) Regulation Mechanisms and Roles in Antiviral Innate Immune Responses. J. Zhejiang Univ. Sci. B 2021, 22, 609.
- Molteni, M.; Gemma, S.; Rossetti, C. The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediators Inflamm. 2016, 2016, 6978936.
- Zhang, Y.; Liang, X.; Bao, X.; Xiao, W.; Chen, G. Toll-like Receptor 4 (TLR4) Inhibitors: Current Research and Prospective. Eur. J. Med. Chem. 2022, 235, 114291.
- Caballero, I.; Boyd, J.; Alminanã, C.; Sánchez-López, J.A.; Basatvat, S.; Montazeri, M.; Maslehat Lay, N.; Elliott, S.; Spiller, D.G.; White, M.R.H.; et al. Understanding the Dynamics of Toll-like Receptor 5 Response to Flagellin and Its Regulation by Estradiol. Sci. Rep. 2017, 7, 40981.
- Zhang, W.; Wang, L.; Sun, X.H.; Liu, X.; Xiao, Y.; Zhang, J.; Wang, T.; Chen, H.; Zhan, Y.Q.; Yu, M.; et al. Toll-like Receptor 5-Mediated Signaling Enhances Liver Regeneration in Mice. Mil. Med. Res. 2021, 8, 16.
- Kim, J.H.; Kordahi, M.C.; Chac, D.; William DePaolo, R. Toll-like Receptor-6 Signaling Prevents Inflammation and Impacts Composition of the Microbiota during Inflammation-Induced Colorectal Cancer. Cancer Prev. Res. 2020, 13, 25–40.
- De Almeida, L.A.; Macedo, G.C.; Marinho, F.A.V.; Gomes, M.T.R.; Corsetti, P.P.; Silva, A.M.; Cassataro, J.; Giambartolomei, G.H.; Oliveira, S.C. Toll-like Receptor 6 Plays an Important Role in Host Innate Resistance to Brucella Abortus Infection in Mice. Infect. Immun. 2013, 81, 1654–1662.
- Bortolotti, D.; Gentili, V.; Rizzo, S.; Schiuma, G.; Beltrami, S.; Strazzabosco, G.; Fernandez, M.; Caccuri, F.; Caruso, A.; Rizzo, R. TLR3 and TLR7 RNA Sensor Activation during SARS-CoV-2 Infection. Microorganisms 2021, 9, 1820.
- Macedo, A.B.; Novis, C.L.; de Assis, C.M.; Sorensen, E.S.; Moszczynski, P.; Huang, S.H.; Ren, Y.; Spivak, A.M.; Jones, R.B.; Planelles, V.; et al. Dual TLR2 and TLR7 Agonists as HIV Latency-Reversing Agents. JCI Insight 2018, 3.
- De Marcken, M.; Dhaliwal, K.; Danielsen, A.C.; Gautron, A.S.; Dominguez-Villar, M. TLR7 and TLR8 Activate Distinct Pathways in Monocytes during RNA Virus Infection. Sci. Signal. 2019, 12, eaaw1347.
- Salvi, V.; Nguyen, H.O.; Sozio, F.; Schioppa, T.; Gaudenzi, C.; Laffranchi, M.; Scapini, P.; Passari, M.; Barbazza, I.; Tiberio, L.; et al. SARS-CoV-2-Associated SsRNAs Activate Inflammation and Immunity via TLR7/8. JCI Insight 2021, 6, e150542.
- Costa, T.J.; Potje, S.R.; Fraga-Silva, T.F.C.; da Silva-Neto, J.A.; Barros, P.R.; Rodrigues, D.; Machado, M.R.; Martins, R.B.; Santos-Eichler, R.A.; Benatti, M.N.; et al. Mitochondrial DNA and TLR9 Activation Contribute to SARS-CoV-2-Induced Endothelial Cell Damage. Vascul. Pharmacol. 2022, 142, 106946.
- Khan, N.S.; Lukason, D.P.; Feliu, M.; Ward, R.A.; Lord, A.K.; Reedy, J.L.; Ramirez-Ortiz, Z.G.; Tam, J.M.; Kasperkovitz, P.V.; Negoro, P.E.; et al. CD82 Controls CpG-Dependent TLR9 Signaling. FASEB J. 2019, 33, 12500–12514.
- Lester, S.N.; Li, K. Toll-Like Receptors in Antiviral Innate Immunity. J. Mol. Biol. 2014, 426, 1246.
- Gaglia, M.M. Anti-Viral and pro-Inflammatory Functions of Toll-like Receptors during Gamma-Herpesvirus Infections. Virol. J. 2021, 18, 218.
- Shah, M.; Anwar, M.A.; Kim, J.H.; Choi, S. Advances in Antiviral Therapies Targeting Toll-like Receptors. Expert Opin. Investig. Drugs 2016, 25, 437–453.
- Vijay, K. Toll-like Receptors in Immunity and Inflammatory Diseases: Past, Present, and Future. Int. Immunopharmacol. 2018, 59, 391.
- Federico, S.; Pozzetti, L.; Papa, A.; Carullo, G.; Gemma, S.; Butini, S.; Campiani, G.; Relitti, N. Modulation of the Innate Immune Response by Targeting Toll-like Receptors: A Perspective on Their Agonists and Antagonists. J. Med. Chem. 2020, 63, 13466–13513.
- O’Neill, L.A.J.; Hennessy, E.J.; Parker, A.E. Targeting Toll-like Receptors: Emerging Therapeutics? Nat. Rev. Drug Discov. 2010, 9, 293–307.
- Zheng, W.; Xu, Q.; Zhang, Y.; Xiaofei, E.; Gao, W.; Zhang, M.; Zhai, W.; Rajkumar, R.S.; Liu, Z. Toll-like Receptor-Mediated Innate Immunity against Herpesviridae Infection: A Current Perspective on Viral Infection Signaling Pathways. Virol. J. 2020, 17, 192.
- Boehme, K.W.; Compton, T. Innate Sensing of Viruses by Toll-Like Receptors. J. Virol. 2004, 78, 7867.
- Ge, Y.; Mansell, A.; Ussher, J.E.; Brooks, A.E.S.; Manning, K.; Wang, C.J.H.; Taylor, J.A. Rotavirus NSP4 Triggers Secretion of Proinflammatory Cytokines from Macrophages via Toll-Like Receptor 2. J. Virol. 2013, 87, 11160–11167.
- Dela Justina, V.; Giachini, F.R.; Priviero, F.; Webb, R.C. COVID-19 and Hypertension: Is There a Role for DsRNA and Activation of Toll-like Receptor 3? Vascul. Pharmacol. 2021, 140, 106861.
- Lau, Y.F.; Tang, L.H.; Ooi, E.E. A TLR3 Ligand That Exhibits Potent Inhibition of Influenza Virus Replication and Has Strong Adjuvant Activity Has the Potential for Dual Applications in an Influenza Pandemic. Vaccine 2009, 27, 1354–1364.
- Miller, R.L.; Meng, T.C.; Tomai, M.A. The Antiviral Activity of Toll-like Receptor 7 and 7/8 Agonists. Drug News Perspect. 2008, 21, 69–87.
- Rose, W.A.; McGowin, C.L.; Pyles, R.B. FSL-1, a Bacterial-Derived Toll-like Receptor 2/6 Agonist, Enhances Resistance to Experimental HSV-2 Infection. Virol. J. 2009, 6, 195.
- Bowie, A.G.; Unterholzner, L. Viral Evasion and Subversion of Pattern-Recognition Receptor Signalling. Nat. Rev. Immunol. 2008, 8, 911–922.
- Duan, S.; Xu, X.; Wang, J.; Huang, L.; Peng, J.; Yu, T.; Zhou, Y.; Cheng, K.; Liu, S. TLR1/2 Agonist Enhances Reversal of HIV-1 Latency and Promotes NK Cell-Induced Suppression of HIV-1-Infected Autologous CD4 + T Cells. J. Virol. 2021, 95, e00816-21.
- Girkin, J.; Loo, S.L.; Esneau, C.; Maltby, S.; Mercuri, F.; Chua, B.; Reid, A.T.; Veerati, P.C.; Grainge, C.L.; Wark, P.A.B.; et al. TLR2-Mediated Innate Immune Priming Boosts Lung Anti-Viral Immunity. Eur. Respir. J. 2021, 58, 2001584.
- Howell, M.D.; Gallo, R.L.; Boguniewicz, M.; Jones, J.F.; Wong, C.; Streib, J.E.; Leung, D.Y.M. Cytokine Milieu of Atopic Dermatitis Skin Subverts the Innate Immune Response to Vaccinia Virus. Immunity 2006, 24, 341–348.
- Hutchens, M.A.; Luker, K.E.; Sonstein, J.; Núñez, G.; Curtis, J.L.; Luker, G.D. Protective Effect of Toll-like Receptor 4 in Pulmonary Vaccinia Infection. PLoS Pathog. 2008, 4, e1000153.
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of Double-Stranded RNA and Activation of NF-KappaB by Toll-like Receptor 3. Nature 2001, 413, 732–738.
- Mazaleuskaya, L.; Veltrop, R.; Ikpeze, N.; Martin-Garcia, J.; Navas-Martin, S. Protective Role of Toll-like Receptor 3-Induced Type I Interferon in Murine Coronavirus Infection of Macrophages. Viruses 2012, 4, 901–923.
- Zhou, Y.; Wang, X.; Liu, M.; Hu, Q.; Song, L.; Ye, L.; Zhou, D.; Ho, W. A Critical Function of Toll-like Receptor-3 in the Induction of Anti-Human Immunodeficiency Virus Activities in Macrophages. Immunology 2010, 131, 40–49.
- Isogawa, M.; Robek, M.D.; Furuichi, Y.; Chisari, F.v. Toll-like Receptor Signaling Inhibits Hepatitis B Virus Replication in Vivo. J. Virol. 2005, 79, 7269–7272.
- Guillot, L.; le Goffic, R.; Bloch, S.; Escriou, N.; Akira, S.; Chignard, M.; Si-Tahar, M. Involvement of Toll-like Receptor 3 in the Immune Response of Lung Epithelial Cells to Double-Stranded RNA and Influenza A Virus. J. Biol. Chem. 2005, 280, 5571–5580.
- Zhang, M.; Yan, Z.; Wang, J.; Yao, X. Toll-like Receptors 7 and 8 Expression Correlates with the Expression of Immune Biomarkers and Positively Predicts the Clinical Outcome of Patients with Melanoma. Onco Targets Ther. 2017, 10, 4339.
- Drake, M.G.; Evans, S.E.; Dickey, B.F.; Fryer, A.D.; Jacoby, D.B. Toll-like Receptor-2/6 and Toll-like Receptor-9 Agonists Suppress Viral Replication but Not Airway Hyperreactivity in Guinea Pigs. Am J Respir Cell Mol. Biol. 2013, 48, 790–796.
- Proud, P.C.; Tsitoura, D.; Watson, R.J.; Chua, B.Y.; Aram, M.J.; Bewley, K.R.; Cavell, B.E.; Cobb, R.; Dowall, S.; Fotheringham, S.A.; et al. Prophylactic Intranasal Administration of a TLR2/6 Agonist Reduces Upper Respiratory Tract Viral Shedding in a SARS-CoV-2 Challenge Ferret Model. EBioMedicine 2021, 63, 103153.
- Lau, Y.F.; Tang, L.H.; Ooi, E.E.; Subbarao, K. Activation of the Innate Immune System Provides Broad-Spectrum Protection against Influenza A Viruses with Pandemic Potential in Mice. Virology 2010, 406, 80–87.
- Christopher, M.; Wong, J. Use of Toll-Like Receptor 3 Agonists Against Respiratory Viral Infections. Antiinflamm. Antiallergy Agents Med. Chem. 2011, 10, 327.
- Kim, M.; Lee, J.E.; Cho, H.; Jung, H.G.; Lee, W.; Seo, H.Y.; Lee, S.H.; Ahn, D.G.; Kim, S.J.; Yu, J.W.; et al. Antiviral Efficacy of Orally Delivered Neoagarohexaose, a Nonconventional TLR4 Agonist, against Norovirus Infection in Mice. Biomaterials 2020, 263, 120391.
- Luchner, M.; Reinke, S.; Milicic, A. TLR Agonists as Vaccine Adjuvants Targeting Cancer and Infectious Diseases. Pharmaceutics 2021, 13, 142.
- Georgel, A.F.; Cayet, D.; Pizzorno, A.; Rosa-Calatrava, M.; Paget, C.; Sencio, V.; Dubuisson, J.; Trottein, F.; Sirard, J.C.; Carnoy, C. Toll-like Receptor 5 Agonist Flagellin Reduces Influenza A Virus Replication Independently of Type I Interferon and Interleukin 22 and Improves Antiviral Efficacy of Oseltamivir. Antiviral Res. 2019, 168, 28–35.
- Melin, N.; Sánchez-Taltavull, D.; Fahrner, R.; Keogh, A.; Dosch, M.; Büchi, I.; Zimmer, Y.; Medová, M.; Beldi, G.; Aebersold, D.M.; et al. Synergistic Effect of the TLR5 Agonist CBLB502 and Its Downstream Effector IL-22 against Liver Injury. Cell Death Dis. 2021, 12, 366.
- SenGupta, D.; Brinson, C.; DeJesus, E.; Mills, A.; Shalit, P.; Guo, S.; Cai, Y.; Wallin, J.J.; Zhang, L.; Humeniuk, R.; et al. The TLR7 Agonist Vesatolimod Induced a Modest Delay in Viral Rebound in HIV Controllers after Cessation of Antiretroviral Therapy. Sci. Transl. Med. 2021, 13, eabg3071.
- Vanwalscappel, B.; Tada, T.; Landau, N.R. Toll-like Receptor Agonist R848 Blocks Zika Virus Replication by Inducing the Antiviral Protein Viperin. Virology 2018, 522, 199–208.
- Amin, O.E.; Colbeck, E.J.; Daffis, S.; Khan, S.; Ramakrishnan, D.; Pattabiraman, D.; Chu, R.; Micolochick Steuer, H.; Lehar, S.; Peiser, L.; et al. Therapeutic Potential of TLR8 Agonist GS-9688 (Selgantolimod) in Chronic Hepatitis B: Remodeling of Antiviral and Regulatory Mediators. Hepatology 2021, 74, 55–71.