Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Cristian Tudor Cozma.
The renin-angiotensin-aldosterone system (RAAS) is comprised of two main branches, with ACE2 representing a crucial component of the protective part of the complex. The ACE2/Ang (1-7) axis is well represented in the testis, heart, brain, kidney, and intestine. Infection with the novel SARS-CoV-2 virus determines downregulation of ACE2 and interrupts the equilibrium between ACE and ACE2 in these organs.
- renin-angiotensin-aldosterone system
- RAAS
- angiotensin-converting enzyme-2
- ACE2
- angiotensin 1-7
- Ang (1-7)
1. Introduction
Angiotensin-converting enzyme-2 (ACE2), a type I transmembrane protein expressed in many organs such as the heart, kidneys, lungs (type II alveolar cell), and intestine, plays a central role in the pathology of COVID-19 infection [1]. As it was in the case of SARS-CoV, the coupling of the virus molecule to the ACE2 receptor leads to fusion and downregulation [2,3][2][3]. Angiotensin-converting enzyme (ACE) and ACE2 are involved in the function regulation of several organs, metabolic condition, and, ultimately, the renin-angiotensin-aldosterone system (RAAS). Currently, this axis is described as two closely interlocking components: a deleterious arm—ACE—and a protective arm—ACE2 [4]. Despite the beneficial side of ACE2, a higher propensity for severe forms of infection has been previously assigned to higher levels of circulating ACE2 [5]. The vast majority of the effects precipitated by COVID-19 infection on key peripheral organs are linked to derangement of the ACE2/ACE axis. There is an ever-growing number of studies focused on observing the impact of the infection outside the lung. Additionally, the considerable amount of medical records allows for a thorough analysis of risk factors.
2. RAAS—The Classical View
The classical view of the renin-angiotensin-aldosterone system considers three peptides whose names comprise the acronym RAA axis. Angiotensinogen is converted to angiotensin I by renin, then further to Ang II by ACE.
Conventionally, the receptors angiotensin II binds to are only angiotensin receptor (AT) type-1 (AT1-R) and AT2-R. There have been other subtypes reported, such as AT3-R and AT4-R; however, the former has not been assigned a gene, so its existence is uncertain, while the latter is part of the AT4-R/Ang IV yet poses no affinity towards Ang II or its analogues (Figure 1) [6].
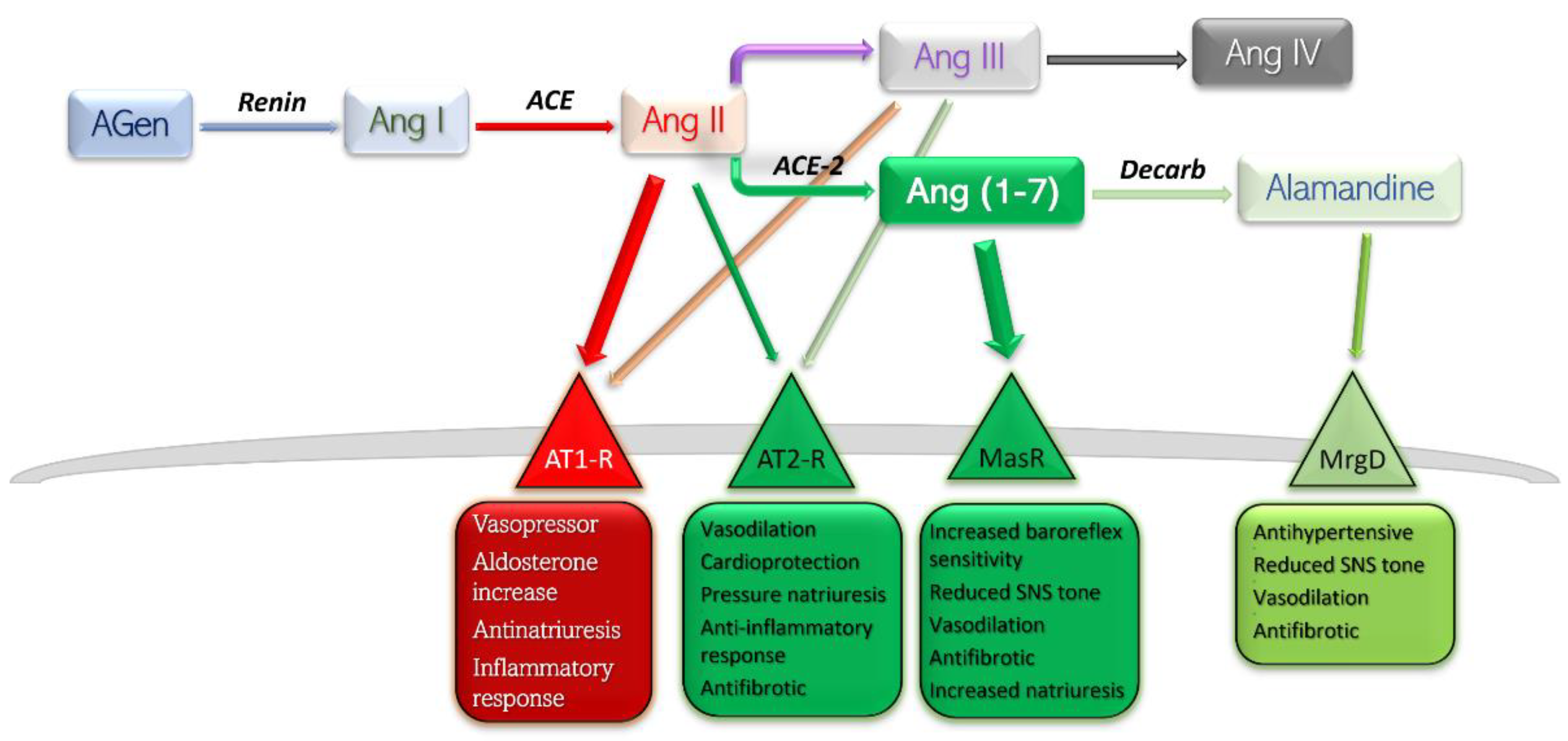
Figure 1. Schematic representation of RAAS components, their main receptors, and main actions. Deleterious side is shown in red-rimmed boxes; protective side is shown in green-rimmed boxes. Abbreviations: Agen, angiotensinogen; Ang, angiotensin; ACE, angiotensin-converting enzyme; Decarb, unspecified decarboxylases; MasR, Mas receptor pathway; MrgD, Mas-related G protein coupled receptor; AT, angiotensin receptor.
Currently, RAAS is viewed as a balance between, on one side, the detrimental actions of Ang II—AT1-R, and, on the other side, the protective effects of Ang II—AT2-R, Ang (1-7)—Mas receptor pathway, and Alamandine—Mas-related G protein receptor pathway. Stimulation of AT1-R subtype by the main peptide driver of RAAS—Ang II—leads to vasoconstriction, antinatriuresis, aldosterone level increase, sympathetic nervous system upregulation, and inflammation-mediated organ damage. Stimulation of AT2-R is generally considered to yield opposite effects, and is therefore protective [6]. The effects mediated by Ang (1-7)—MasR and Alamandine—MrgD are thoroughly discussed further in this text.
The degree of independence between the AT1 and AT2 receptors is highlighted by in vivo consequences after angiotensin-receptor blockers (ARB) administration [7]. Selective blockade of AT1-R would lead to a proportionally higher quantity of substrate binding to the remaining substrate, AT2-R. Furthermore, blockade of AT1-R at renal level results in increased levels of circulating renin and therefore higher levels of circulating Ang II, which would exhibit its protective effects on the only free receptors, AT2. Several studies have reported dramatically increased effects of AT2-R selective stimulation in the presence of small-dose ARB, which may be explained by a higher constitutive expression of AT1-R compared to AT2-R [8,9,10][8][9][10].
Metabolism end-products of Ang are Ang III and Ang IV. The former has a half-life five times lower in plasma than Ang II, and the latter exhibits its effects largely at the central level, with very low influence on peripheral AT1/2 subtypes [11]. For these reasons, Ang III and Ang IV contribute only slightly to the whole array of effects of Ang II.
3. RAAS—The Alternative Pathway
A new involvement in blood pressure control was unveiled in the early 2000s [12,13][12][13]. In this axis, Ang II is converted to Ang (1-7) by the catalytic activity of the then novel discovery, ACE2. The ACE–Ang II–AT1-R axis hyperactivation commonly leads to deleterious effects such as vasoconstriction, inflammation, endothelial dysfunction, thrombosis, and the well-established pro-hypertensive profile; the other, ACE2–Ang (1-7)–MasR typically reverts the forementioned effects. Ang (1-7) is found at a rather pseudo-stationary concentration state. There are at least three enzymes which aid in synthesis of Ang (1-7), and the reaction with the highest rate is different among the peripheral organs [14] (Figure 2).
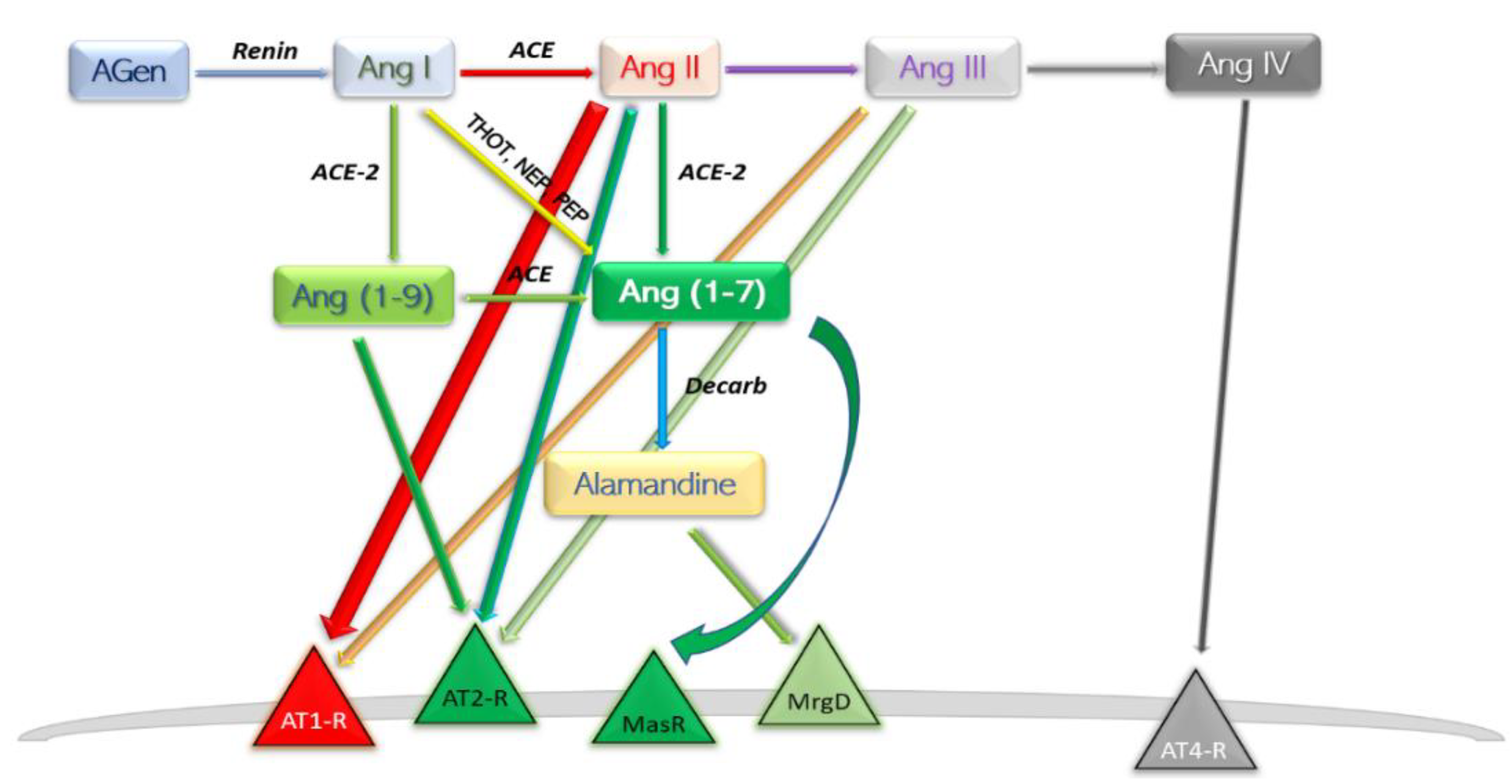
Figure 2. Schematic depiction of the modern view ACE2 axis and its products. AGen, angiotensinogen; Ang, angiotensin; ACE, angiotensin-converting enzyme; Decarb, unspecified aspartate decarboxylases; THOT, thimet oligopeptidase; NEP, neutral endopeptidase; PEP, prolyl endopeptidase; MasR, Mas axis; MrgD, Mas-related G protein receptor; AT–R, angiotensin receptor subtype.
3.1. ACE2 Enzyme
Following the efforts of discovering “ACE-like enzymes” in Drosophila melanogaster, mammalian homologue of ACE has been cloned and named “angiotensin-converting enzyme 2” (ACE2) [12,13,15][12][13][15]. ACE2 is an extracellular transmembrane protein expressed more restrictively to the surface of specific tissues including heart, kidney, endothelium, and testis [13]. Like ACE, it can be cleaved from the cell surface, resulting in the soluble form sACE2 [16]. Despite exhibiting considerable similarity and identity to human ACE, ACE2 functions exclusively as a carboxypeptidase, cleaving only the C-terminal residue from Ang I, yielding Ang (1-9) and from Ang II, likewise yielding Ang (1-7). Regardless, both the structures of ACE2 and ACE display a zinc-binding motif; typical angiotensin-converting enzyme inhibitors (ACEI) do not return any inhibition on ACE2 activity [12]. Arguably, inhibitors designed to act on C-terminal dipeptide-releasing enzymes—ACE—do not bind efficiently to C-terminal cleaving enzymes—ACE2.
3.2. SARS-CoV-2 and Its Receptor, ACE2
A full-fledged crisis emerged as the severe acute respiratory syndrome virus 2 (SARS-CoV-2) pandemics took over in late 2019. Most coronaviruses determine moderate enteric and pulmonary distress through the establishment of a proinflammatory profile [17]. SARS-Cov-2 belongs to the genus Betacoronavirus, but unlike the common coronaviruses, precipitates serious respiratory dysfunction [18,19][18][19]. ACE2 behaves as a receptor for SARS-CoV-2, and once the complex is formed, it is immediately internalized [20,21][20][21]. Surface expression of ACE2 is therefore diminished through consumption; circulating serum levels—sACE2—may also decrease, accounting for an absolute decrease in shedding. A decline in ACE2 levels will most probably be denoted by an alteration in synthesis of both Ang (1-9) and Ang (1-7). Furthermore, degradation of the hypertensive components of RAAS may be impeded by the low levels of enzyme ACE2. Therefore, a disequilibrium throughout the entire RAA axis is expected. While Ang (1-9) exhibits only few of the characteristics SARS-CoV-2 interferes with, Ang (1-7) is the most important substrate to account for the extensive effects of the infection.
3.3. Angiotensin (1-9)
Ang (1-9) is considered an intermediate product. The production step is catalysed by ACE-2, while consumption is catalysed by ACE. Ang (1-9) should exhibit in vivo a pseudo-stationary concentration; the absolute value of concentration at a specific time is directly determined by—amongst other factors—the ratio of the enzymatic activities of ACE-2 and ACE. Any alteration in ACE-2 activity would therefore yield significant effects on a systemic scale. The first direct receptor-mediated effect turned out to be the antihypertrophic effect mediated by AT2-R, independent of MasR pathway manipulation [22].
Infusion of Ang (1-9) in hypertensive elevated Ang II level rats resulted in significant blood pressure drop and circulating Ang II level reduction. A co-infusion experiment aimed to disclose the receptor for Ang (1-9) proved that MasR antagonist did not affect the beneficial properties of Ang (1-9), while AT2-R antagonist completely reversed the effects [22]. Controversial claims regarding either a possible interaction of Ang (1-9) with AT1-R, or in vivo transformation of this peptide to Ang II, were advanced in an attempt to explain the in vivo arterial pro-thrombotic activity exhibited by Ang (1-9) in rats [23,24][23][24]. To date, there are no reports of an enzyme to aid in conversion of Ang (1-9) to Ang II, nor a quantified affinity towards receptor subtypes other than AT2-R angiotensin. Extracellular signal-regulated kinase (ERK)1/2 pathway downregulation seems to be the key mechanism involved in the antifibrotic and antihypertrophic properties exhibited through AT2-R stimulation [25].
3.4. Angiotensin (1-7)
Angiotensin (1-7) is the most potent product of the protective arm of RAAS. Synthesis can take place three ways: (a) directly and most efficient, from Ang II by catalytic action of ACE2 [26]; (b) directly, from Ang I, catalysed by thimet oligopeptidase (THOT), neutral endopeptidase (NEP), and prolyl endopeptidase (PEP) [27,28][27][28]; and (c) through an intermediary product Ang (1-9), and therefore with a slow reaction speed, by means of ACE2 then ACE [29]. Degradation of Ang (1-7) takes place by parallel first-order kinetic reactions; decarboxylation yields a novel compound—Alamandine—by a not currently individualised enzyme. Interaction of alamandine with Mas-related G protein-coupled receptor (MrgD) explained some of the previously noted effects of Ang (1-7) which could not be assigned to Mas axis [30].
3.4.1. Intracellular Signalling Pathways Employed by Angiotensin (1-7)
The majority of the effects generated by this heptapeptide are mediated by the MasR pathway. Downstream activation leads to Akt phosphorylation, upregulation of inducible nitric oxide synthases, and intracellular cyclic guanosine monophosphate (GMP) increase.
Besides the classical MasR pathway, Ang (1-7) mediates some of its cardioprotective effects by behaving as a beta-arrestin recruiter. Agonistic action at the active site of AT1-R leads to recruitment of beta1/2 arrestins, intracellular molecules which effectively block the active site in an irreversible manner. AT1-R engagement by Ang (1-7) leads ultimately to a transient activation of ERK1/2 axis, displaying a cardioprotective profile [31]. The affinity towards receptors other than MasR was also independently demonstrated by using human kidney (HK-2) cells [32]. Effects of low-concentration MasR agonist were not influenced by subsequent MasR blockade, but reversed by AT2 and AT1-R blockade, which owes a degree of affinity of Ang (1-7) to both receptors (Figure 3).
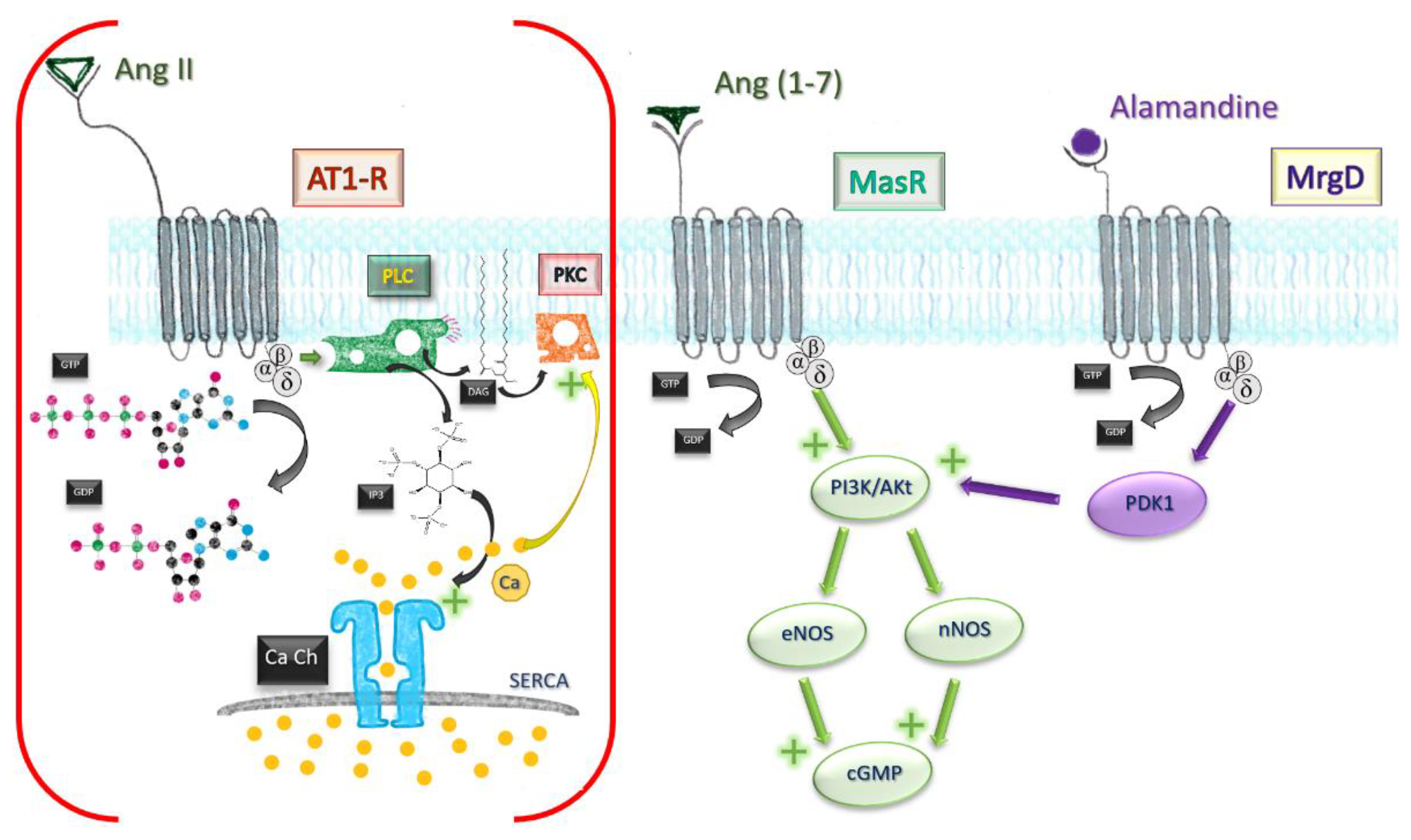
Figure 3. Comparative mechanisms of Ang II and Ang (1-7). Ang, angiotensin; MrgD, Mas-related G protein receptor D; AT1-R, angiotensin receptor type I; GTP, guanosine triphosphate; GDP, guanosine diphosphate; PLC, phospholipase C; PKC, protein kinase C; DAG, diacylglycerol; IP3, triphosphate inositol; Ca Ch, Calcium channel of smooth endoplasmic reticulum; eNOS, endothelial nitric oxide synthase; nNOS, neuronal; PI3K/Akt pathway; PDK1, phosphoinositide-dependent protein kinase 1.
Influence on Metabolism
Activation of Glycogen Synthase Kinase-3 beta (GSK3β), a crucial insulin mediator, may be Akt-dependent or direct; nevertheless, metabolic effects should be expected after manipulation of Ang (1-7), and indeed, oral administration of coated peptide attenuates hyperglycaemia in type 2 diabetes mellitus rodents showing impaired insulin sensitivity [33,34][33][34]. While GSK3β further downstream controls glycogen synthesis, phosphorylation of Akt Substrate-160 (AS160) by Akt is required for Glucose Transporter Type-4 translocation to occur. GSK3β and AS160 synergically inhibit phosphorylation of the inhibitory site at Insulin Receptor Substrate 1.
ACE2 knockout mice showed reduced concentration of tryptophan and other large amino acids in blood (valine, threonine, and tyrosine) [35]. Histologically, the mice displayed diffuse mucosal inflammation and altered intestinal microbiota. Inflammatory susceptibility in ACE2-deficient subjects was remedied by supplying only tryptophan, which may indicate a benefit in a tryptophan rich diet in COVID patients. Along with the current knowledge regarding blood sugar levels and microbiota, insulin resistance and impaired glucose homeostasis can be partially explained by an imbalance of ACE forms [36].
Oxidative Stress
Nicotinamide adenine dinucleotide phosphate (NADPH) inhibition leads to reduced oxidative stress by inhibiting the detrimental activity of inducible nitric oxide (NO) synthase [37]. ACE2 knockout display hypertensive phenotypes aggravated by endothelial dysfunction, proved by the worse outcome induced by diabetes- and shock-induced kidney injury, viral lung injury, and chronic liver injury [38]. The unifying characteristic among these models is an increase in oxidative stress in ACE2 knockout male mice [39,40][39][40].
Arterial Blood Pressure
Vascular smooth muscle tone may be modulated either directly—as determined by upregulation of endothelial and neuronal NO synthase, subsequent cyclic GMP rise and vasodilation—or indirectly. Ang 1-7 treatment leads to normalisation of surface expression of the proteins Tumor Necrosis Factor (TNF)-Alpha Converting Enzyme (TACE) and Sodium Hydrogen Exchanger-3 (NHE3) in proximal renal tubule cells [41]. TACE is responsible for surface ACE-2 shedding; the high levels expressed in hypertension only compound the problem regarding low activity of Ang 1-7 [42]. NHE3 is a key transporter in proximal renal tubule cells; upregulation leads to increased reabsorption and drop in diuresis [43]. The reactive oxidative species (ROS) and NO generation in genetic deletion of ACE2 enabled researchers to observe the direct effect of Ang (1-7), and only slight but consistent elevation of blood pressure was noted [39]. Results are, however, discordant among different genetic mouse strains [44]. Vascular dysfunction—loss of the vessel ability to modulate local tone—only reinforces the previously noted consistently increased vascular tone [41].
Apoptosis
Mitogen-activated protein kinase pathway and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) transcription factor are both toned down in vascular smooth muscle cells, endothelium, and proximal tubule in kidney after infusion of the heptapeptide [45[45][46],46], resulting in a lesser inflammatory response, reduced cell death, reduced fibrosis, and satisfactory preservation of organ function. Finally, stimulation of MrgD by alamandine may account for the effects observed in triple blockade (MasR, AT1, AT2-R). Through phosphoinositide-dependent protein kinase 1 (PDK1), it mediates phosphorylation and activation of Akt, which downstream will activate inducible NO synthases. Figure 4 sums up the most important paths Ang (1-7) acts.
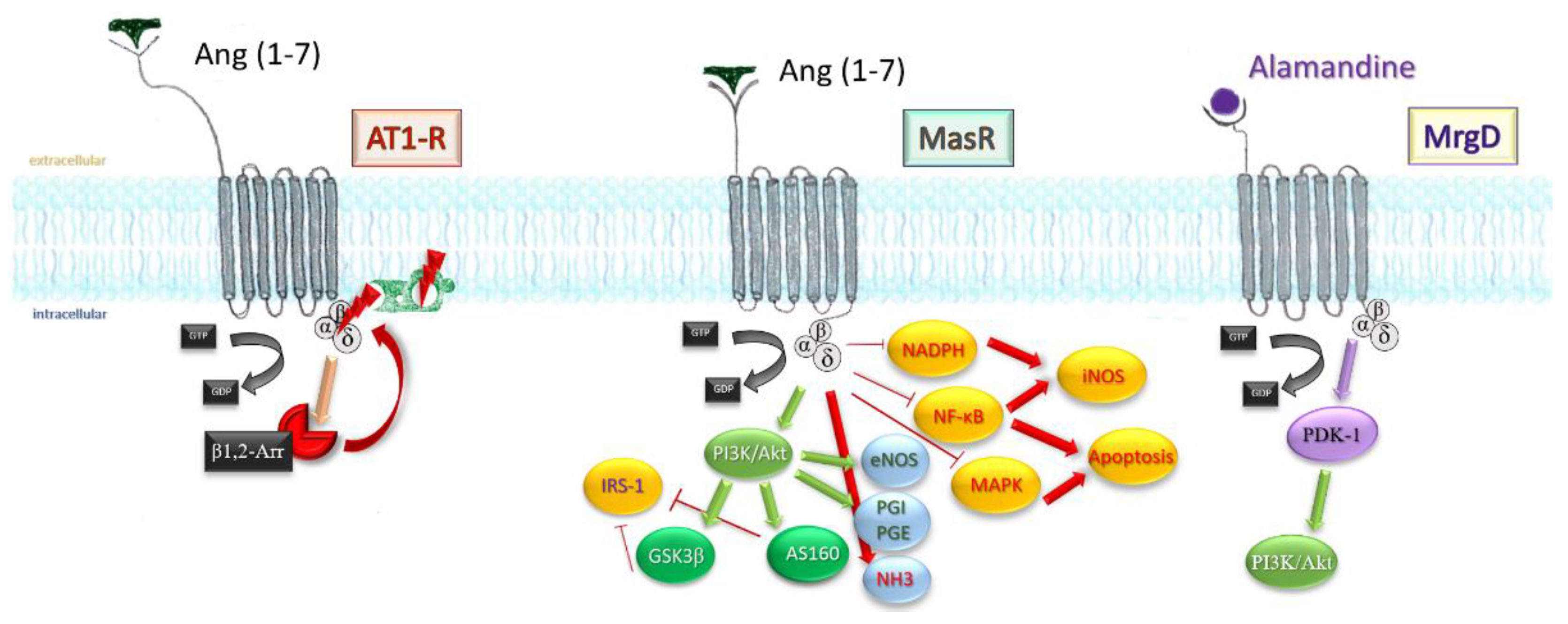
Figure 4. An extensive scheme displaying the mechanisms Ang (1-7) influences on an intracellular level. β-arr1/2, β-arrestins; GSK3β, glycogen synthase kinase 3β; AS160, Akt substrate 160; IRS-1, insulin receptor substrate 1; PGI, PGE, prostaglandins; NHE3, sodium hydrogen exchanger 3; NF-KB, nuclear transcription factor; iNOS, inducible nitrous oxide synthase; MAPK, mithogen-activated protein kinase; NADPH, nicotin-amide-diphospho-hydrogenase.
3.4.2. Effects Mediated by Angiotensin (1-7) in Specific Organs
In order to assess the effects mediated by Ang (1-7) in an individual setting, several strategies have been employed to manipulate either the substrate or the receptor activities: pharmacological concentration alterations of circulating Ang (1-7), genetically modified expression of ACE2, and MasR. Significantly involved in the Ang (1-7)/MasR axis due to the high local Mas expression, the brain exhibits both local and systemic effects in response to manipulation of this axis.
Brain
Metabolism of the heptapeptide closely resembles the description in Figure 2. All components of RAS described above have been determined in brain tissue; however, not every compartment exhibits all of them, nor has any neuron type been identified to produce all those substances [47]. Despite that, Ang (1-7) presence has only been selectively demonstrated; ACE2 is present through every single brain compartment, mainly in neurons rather than glia cells [48]. MasR was identified in certain compartments mostly—but not exclusively—related to cardiovascular control: nucleus tract solitary (NTS), rostral and caudal ventrolateral medulla (RVLM, CVLM), paraventricular nucleus (PVN), and supraoptic nucleus (SON) [49].
Improved modulation of baroreflex by enhancing the bradycardic component, cardiac autonomic balance regaining, and renal sympathetic tone control restauration stand as explanation for the experiment in which long-term stimulation of Ang (1-7) axis leads to significant drop in hypertensive response to Aldosterone-NaCl artificial infusions or high-salt regime [50,51][50][51]. Aldosterone-NaCl treatment mimics the effect of Ang II systemically, while also downregulating any intrinsic renin or Ang II secretion. Deoxycorticosterone acetate (DOCA)-salt hypertension is predominantly characterized by upregulated sympathetic activity. Mas knockout mice (equivalent to ACE imbalanced profile) displayed—besides impaired baroreflex and aberrant renal sympathetic tone activity rise—blunted Jarisch-Bezold and chemoreceptor reflexes, with the entire neural network for blood pressure control being influenced [52].
The baroreflex response is the most intricate of all previously mentioned cardiovascular mechanisms. ACE inhibition led to improvement of bradycardic component in normotensive and spontaneous hypertensive rats (SHR). Inhibition of ACE2 axis by infusion of A-779 (an antagonist of Ang (1-7)) led to a significantly depressed sensitivity in normotensive, yet to a mild, almost insignificant attenuation of baroreflex in SHR, which comes to emphasize the imbalance between the two Angiotensin axis in the hypertensive subjects [53]. Infusion of A-779 in ACE inhibitor-treated SHR subjects promptly reverted the beneficial effects [54]. The favourable outcome met by ACEI treatment was most likely exhibited through Ang (1-7). Increased levels of Ang (1-7) at NTS—a key cardiovascular reflex command centre—and signalling through Phosphoinositide 3-kinase (PI3K) pathway may be responsible [55].
Fructose-fed rats treated with intracerebroventricular high-dose Ang (1-7) displayed protective (High-density lipoprotein) HDL-Cholesterol values, normal glucose tolerance, and normal levels of insulin. The peripheral cardioprotective and metabolic effects of central administration of Ang (1-7) were mediated via shifting Ang II/ACE2 equilibrium and further consequences on NO production rise (neuronal NOS activation via Mas axis) [56,57][56][57].
Emotional stress and anxiety-like syndromes were noted in Mas-deficient animals [58]. Chronic intracerebroventricular infusion of Ang (1-7) proved effective in reducing both anxiety- and depression-like syndromes [59]. Increase in ACE2 activity by use of ACEI showed a mood improvement in depression-suffering hypertensive patients [60]. The emotional response to stress can therefore be improved by administering ACEI class drugs, discontinuation of which would be deeply unwise from perspectives of both cardiovascular risk and cerebral involvement. Further benefit comes from the modulation of cardiovascular response evoked by acute emotional stress. Microinjections of Ang (1-7) into the basolateral amygdala—an area of the limbic system—lead to attenuation of pressor reflex in response to acute psychic stress [61].
Cardiac Direct Involvement
Coronary vessels seem to be favourably responsive at nanomolar order of concentration, with higher concentrations exhibiting no or constrictive effect [62].
Cardiomyocytes acutely exposed to Ang (1-7) only display a slight elevation in NO release by activation of endothelial and neuronal nitric oxide synthase (eNOS, nNOS) [63]; chronic exposure leads to significant effects on calcium-handling proteins, with increased expression of Sarcoendoplasmic Reticulum Calcium ATPase-2 (SERCA2), higher Calcium (Ca) transient amplitude and faster Ca uptake. This may explain the beneficial effects of ACE2 axis stimulation in chronic heart failure subjects [64].
Identification of Ang (1-7) and Mas axis in sinoatrial node provides the morphopathological fundamentals for the observed biphasic effect (an excessively high local peptide concentration worsened the prognosis) in administration of different concentration of Ang (1-7), only one order of magnitude apart [65,66][65][66]. Long-term overproduction of Ang (1-7) has proved to reduce cardiac fibrosis in many studies, reducing oxidative stress, autophagy prevention, and reducing mitogen-activated fibroblast proliferation [67,68][67][68]. The main axis involved are Mas-R, Insulin-like Growth Factor-1 Receptor (IGF1R)/PI3K/Akt, and alteration of mitogenic prostaglandin profile [69].
Contradicting its expected cardioprotective effects, overexpression of human ACE2 in mouse heart lead to ventricular tachycardia and sudden death [70]. An explanation may arise from the role of apelin, a peptide involved in a variety of cardiovascular pathological processes. Apelin serves as substrate for ACE2 and as a biomarker in cardiovascular diseases including coronary artery disease, stroke, ischemic heart disease, and infarction [71]. The lower the level of apelin, the worse the prognosis in those subjects. Manipulation of ACE axis towards a deep imbalance in favour of ACE2/Ang (1-7)/MasR may, therefore, prove detrimental in already fragile COVID patients.
Kidney
Inside the kidney water regulation network, Ang (1-7) and antidiuretic hormone (AVP) are involved in a very delicate balance. The effects of Ang 1-7 at central level may not parallel the ones on a peripheral level.
Ang (1-7) effects are distinct among different brain areas. Even in the same cerebral structure, there may be a whole range of distinctive effects owing to certain physiopathological conditions. As such, infusions in the PVN in rodents exhibit different effects according to the state of the subject involved. Following Ang (1-7) microinjection in a healthy subject, an increase in AVP was demonstrated [72]. However, an inverse relationship was noted between Ang (1-7) concentration and AVP release in haemorrhagic conditions of an ethanol-intoxicated model [57]. The underlying mechanism is the already consistent NO-induced release by Ang (1-7), which in the second study displayed an inhibitory effect on AVP secretion.
Ang (1-7) presents a wide array of effects on kidney function, which are way beyond ourthe target. In almost all instances, perfusion/injection of Ang (1-7) in the microenvironment of the studied kidney (or glomerular portion) resulted either in no effect, or natriuresis/diuresis, antiproteinuric, or rarely Ang II antagonism [27]. Antidiuretic effects were noted in only two instances. Worth mentioning is the possible involvement of receptors other than Mas pathway; antidiuretic effects noticed in healthy normotensive rats could be blocked by pre-treatment with Angiotensin Receptor Blockers (ARB) Losartan—Ang (1-7) may display some affinity towards AT1R [73]. Additionally, antidiuretic effect in inner medullary-collecting tubule cells of water-loaded rats seems to involve V2-AVP receptor [74]. Administering A-779 prior to AVP blunted the water reabsorption; similarly, AVP antagonist forskolin prior to Ang (1-7) resulted in no increase in cyclic Adenosine Monophosphate (cAMP) and in no effect in the collecting tubule cell. Cross-antagonist administration following the inspected substance returned no inhibition; the mechanism underlying AVP/Ang (1-7) and their interconnecting substrate-receptor affinity may rely upon binding with subsequent cross-internalization of complex.
Renal vasculature in vitro exhibited afferent arteriole vasodilation in response to Ang (1-7) [75]. Recent in vivo studies prove the vasodilatory effect, yet also the inverse dependence on the degree of RAAS activation [76]; thus, low sodium intake and co-infusion of Ang II lead to hyperactivity of RAAS in the hypertensive human, which diminishes the beneficial effect of intrarenal infusion of Ang (1-7).
Vascular Actions
Ang (1-7) is endogenously produced by vascular endothelium, and therefore induces endothelium-dependent vasorelaxation. Furthermore, it enhances the vasodilator effect of bradykinin in several vascular beds [77].
Besides the well-known effect of vasodilation, Ang (1-7) exhibits antiproliferative and antithrombotic actions [78,79][78][79]. The antiproliferative side is mediated through an increase in prostacyclin (PGI2), which in turn reduced activity of mitogen-activated protein kinase (MAPK) Ang II-stimulated pathway. Neointimal thickness and stenosis reduction in rat stenting model, slowing of the osteogenic transition of vascular smooth muscle cells in calcified specimens, and cardiac fibroblast antiproliferative potential were noted [68,80,81][68][80][81]. The antithrombotic side is mediated by the increase in prostacyclin PGI2 and enhanced release of NO from platelets, both part of the Mas axis [82].
Most of the work around Ang (1-7) has been aimed at understanding its effect on vessels and blood pressure. While Ang II/AT1R is widely distributed in all vascular beds, Ang (1-7)/Mas is specific to certain vascular beds—kidney, lungs, adrenals, brain. In normotensive rats, either short or long-term infusion of Ang (1-7) resulted in increased conductance in the previously mentioned vascular beds. However, a proportional cardiac output increase allowed these subjects to not display any significant blood pressure changes [83].
Many of the vascular effects Ang (1-7) exhibits in vivo are mediated through Mas-R activation, which in turn activates eNOS and enhances NO release. Previously, it is noted also the nNO inducting activity of the same Mas-R. Ang (1-7) is also involved in a fine-tuned network involving PI3K/Akt pathway, as demonstrated by the NO levels in Mas-transfected cells and in vivo transcription activation of Forkhead box protein O1 (FOXO1), a well-known negative regulator of Akt cascade [84,85][84][85]. The same PI3K/Akt pathway is—peripherally—involved in improvement of insulin sensitivity in fructose-fed rats [33].
In human endothelium, Ang (1-7) counteracts effects mediated by Ang II through a common enzyme, Src homology-2 domain-containing protein tyrosine phosphatase-2 (SHP-2). Antioxidant action occurs by opposing the activation of NADPH oxidase by AT1-R, while anti-inflammatory activity occurs through inhibition of NFkB nuclear factor translocation in nuclear cells and attenuation of Vascular Cell Adhesion Molecule-1 (VCAM-1) expression, otherwise an early marker of endothelial dysfunction [86,87][86][87].
Restoration of ACE2 function in stroke-prone spontaneously hypertensive rats (which constitutively present a low ACE2 level) leads to significant stroke risk decrease, lower blood pressure profiles, and improved endothelial function [88,89,90][88][89][90]. Function restoration and perhaps overshooting the imbalance of ACE axis towards the protective side may yield strongly beneficial therapeutic results.
References
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610.
- Freitas, F.C.; Ferreira, P.H.B.; Favaro, D.C.; Oliveira, R.J. Shedding Light on the Inhibitory Mechanisms of SARS-CoV-1/CoV-2 Spike Proteins by ACE2-Designed Peptides. J. Chem. Inf. Model. 2021, 61, 1226–1243.
- Mehrabadi, M.E.; Hemmati, R.; Tashakor, A.; Homaei, A.; Yousefzadeh, M.; Hemati, K.; Hosseinkhani, S. Induced dysregulation of ACE2 by SARS-CoV-2 plays a key role in COVID-19 severity. Biomed. Pharmacoter. 2021, 137.
- Ahmadi Badi, S.; Tarashi, S.; Fateh, A.; Rohani, P.; Masotti, A.; Siadat, S.D. From the Role of Microbiota in Gut-Lung Axis to SARS-CoV-2 Pathogenesis. Mediators Inflamm. 2021, 2021, 6611222.
- Gu, J.; Yin, J.; Zhang, M.; Li, J.; Wu, Y.; Chen, J.; Miao, H. Study on the Clinical Significance of ACE2 and Its Age-Related Expression. J. Inflamm. Res. 2021, 14, 2873–2882.
- Singh, K.D.; Karnik, S.S. Angiotensin Receptors: Structure, Function, Signaling and Clinical Applications. J. Cell. Sign. 2016, 1.
- Carey, R.M. AT2 Receptors: Potential Therapeutic Targets for Hypertension. Am. J. Hypertens. 2017, 30, 339–347.
- Li, X.C.; Widdop, R.E. AT2 receptor-mediated vasodilatation is unmasked by AT1 receptor blockade in conscious SHR. Br. J. Pharmacol. 2004, 142, 821–830.
- Widdop, R.E.; Matrougui, K.; Levy, B.I.; Henrion, D. AT2 receptor-mediated relaxation is preserved after long-term AT1 receptor blockade. Hypertension 2002, 40, 516–520.
- Siragy, H.M.; de Gasparo, M.; Carey, R.M. Angiotensin type 2 receptor mediates valsartan-induced hypotension in conscious rats. Hypertension 2002, 35, 1074–1077.
- Gammelgaard, I.; Wamberg, S.; Bie, P. Systemic effects of angiotensin III in conscious dogs during acute double blockade of the renin-angiotensin-aldosterone-system. Acta Physiol. 2006, 188, 129–138.
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243.
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9.
- Campbell, D.J.; Zeitz, C.J.; Esler, M.D.; Horowitz, J.D. Evidence against a major role for angiotensin converting enzyme-related carboxypeptidase (ACE2) in angiotensin peptide metabolism in the human coronary circulation. J. Hypertens. 2004, 22, 1971–1976.
- Cornell, M.J.; Williams, T.A.; Lamango, N.S.; Coates, D.; Corvol, P.; Soubrier, F.; Hoheisel, J.; Lehrach, H.; Isaac, R.E. Cloning and Expression of an Evolutionary Conserved Single-domain Angiotensin Converting Enzyme from Drosophila melanogaster. J. Biol. Chem. 1995, 270, 13613–13619.
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119.
- Glass, W.G.; Subbarao, K.; Murphy, B.; Murphy, P.M. Mechanisms of host defense following severe acute respiratory syndrome-coronavirus (SARS-CoV) pulmonary infection of mice. J. Immunol. 2004, 173, 4030–4039.
- Kadam, S.B.; Sukhramani, G.S.; Bishnoi, P.; Pable, A.A.; Barvkar, V.T. SARS-CoV-2, the pandemic coronavirus: Molecular and structural insights. J. Basic Microbiol. 2021, 61, 180–202.
- Grasselli, G.; Pesenti, A.; Cecconi, M. Critical Care Utilization for the COVID-19 Outbreak in Lombardy, Italy: Early Experience and Forecast During an Emergency Response. JAMA 2020, 323, 1545–1546.
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574.
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20.
- Ocaranza, M.P.; Moya, J.; Barrientos, V.; Alzamora, R.; Hevia, D.; Morales, C.; Pinto, M.; Escudero, N.; García, L.; Novoa, U.; et al. Angiotensin-(1-9) reverses experimental hypertension and cardiovascular damage by inhibition of the angiotensin converting enzyme/Ang II axis. J. Hypertens. 2014, 32, 771–783.
- Kramkowski, K.; Mogielnicki, A.; Leszczynska, A.; Buczko, W. Angiotensin-(1-9), the product of angiotensin I conversion in platelets, enhances arterial thrombosis in rats. J. Physiol. Pharmacol. 2010, 61, 317–324.
- Mogielnicki, A.; Kramkowski, K.; Hermanowicz, J.M.; Leszczynska, A.; Przyborowski, K.; Buczko, W. Angiotensin-(1-9) enhances stasis-induced venous thrombosis in the rat because of the impairment of fibrinolysis. J. Renin Angiotensin Aldosterone Syst. 2014, 15, 13–21.
- Calò, L.A.; Schiavo, S.; Davis, P.A.; Pagnin, E.; Mormino, P.; D’Angelo, A.; Pessina, A.C. Angiotensin II signaling via type 2 receptors in a human model of vascular hyporeactivity: Implications for hypertension. J. Hypertens. 2010, 28, 111–118.
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F. Hydrolysis of Biological Peptides by Human Angiotensin-converting Enzyme-related Carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843.
- Santos, R.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553.
- Chappell, M.C.; Allred, A.J.; Ferrario, C.M. Pathways of angiotensin-(1-7) metabolism in the kidney. Nephrol. Dial. Transplant. 2001, 16, 22–26.
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51.
- Lautner, R.Q.; Villela, D.C.; Fraga-Silva, R.A.; Silva, N.; Verano-Braga, T.; Costa-Fraga, F.; Jankowski, J.; Jankowski, V.; Sousa, F.; Alzamora, A.; et al. Discovery and characterization of alamandine: A novel component of the renin-angiotensin system. Circ. Res. 2013, 112, 1104–1111.
- Teixeira, L.B.; Parreiras-E-Silva, L.T.; Bruder-Nascimento, T.; Duarte, D.A.; Simões, S.C.; Costa, R.M.; Rodríguez, D.Y.; Ferreira, P.; Silva, C.; Abrao, E.P.; et al. Ang-(1-7) is an endogenous β-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci. Rep. 2017, 7, 11903.
- Patel, S.; Hussain, T. Synergism between Angiotensin receptors ligands: Role of Angiotensin-(1-7) in modulating AT2 R agonist response on nitric oxide in kidney cells. Pharmacol. Res. Perspect. 2020, 8, e00667.
- Muñoz, M.C.; Giani, J.F.; Burghi, V.; Mayer, M.A.; Carranza, A.; Taira, C.A.; Dominici, F.P. The Mas receptor mediates modulation of insulin signaling by angiotensin-(1-7). Regul. Pept. 2012, 177, 1–11.
- Santos, S.H.; Giani, J.F.; Burghi, V.; Miquet, J.G.; Qadri, F.; Braga, J.F.; Todiras, M.; Kotnik, K.; Alenina, N.; Dominici, F.P.; et al. Oral administration of angiotensin-(1-7) ameliorates type 2 diabetes in rats. J. Mol. Med. 2014, 92, 255–265.
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481.
- Niu, M.J.; Yang, J.K.; Lin, S.S.; Ji, X.J.; Guo, L.M. Loss of angiotensin-converting enzyme 2 leads to impaired glucose homeostasis in mice. Endocrine 2008, 34, 56–61.
- Villalobos, L.A.; San Hipólito-Luengo, Á.; Ramos-González, M.; Cercas, E.; Vallejo, S.; Romero, T.; Carraro, R.; Sanchez-Ferrer, C.F.; Peiro, C. The Angiotensin-(1-7)/Mas Axis Counteracts Angiotensin II-Dependent and -Independent Pro-inflammatory Signaling in Human Vascular Smooth Muscle Cells. Front. Pharmacol. 2016, 7.
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.Y.; Al-Omran, M.; Stewart, D.J. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1377–H1384.
- Rabelo, L.A.; Todiras, M.; Nunes-Souza, V.; Qadri, F.; Szijártó, I.A.; Gollasch, M.; Penninger, J.M.; Bader, M.; Santos, R.A.; Alenina, N. Genetic Deletion of ACE2 Induces Vascular Dysfunction in C57BL/6 Mice: Role of Nitric Oxide Imbalance and Oxidative Stress. PLoS ONE 2016, 11, e0150255.
- Wysocki, J.; Ortiz-Melo, D.I.; Mattocks, N.K.; Xu, K.; Prescott, J.; Evora, K.; Ye, M.; Sparks, M.A.; Haque, S.K.; Batlle, D. ACE2 deficiency increases NADPH-mediated oxidative stress in the kidney. Physiol. Rep. 2014, 2, e00264.
- Shi, Y.; Lo, C.S.; Padda, R.; Abdo, S.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S.D. Angiotensin-(1–7) prevents systemic hypertension, attenuates oxidative stress and tubulointerstitial fibrosis, and normalizes renal angiotensin-converting enzyme 2 and Mas receptor expression in diabetic mice. Clin. Sci. 2015, 128, 649–663.
- Zamilpa, R.; Chilton, R.J.; Lindsey, M.L. Tumor necrosis factor-alpha--converting enzyme roles in hypertension-induced hypertrophy: Look both ways when crossing the street. Hypertension 2009, 54, 471–472.
- Knepper, M.A.; Brooks, H.L. Regulation of the sodium transporters NHE3, NKCC2 and NCC in the kidney. Curr. Opin. Nephrol. 2001, 10, 655–659.
- Moritani, T.; Iwai, M.; Kanno, H.; Nakaoka, H.; Iwanami, J.; Higaki, T.; Ishii, E.; Horiuchi, M. ACE2 deficiency induced perivascular fibrosis and cardiac hypertrophy during postnatal development in mice. J. Am. Soc. Hypertens. 2013, 7, 259–266.
- Zhang, F.; Ren, X.; Zhao, M.; Zhou, B.; Han, Y. Angiotensin-(1-7) abrogates angiotensin II-induced proliferation, migration and inflammation in VSMCs through inactivation of ROS-mediated PI3K/Akt and MAPK/ERK signaling pathways. Sci. Rep. 2016, 6, 34621.
- Gava, E.; Samad-Zadeh, A.; Zimpelmann, J.; Bahramifarid, N.; Kitten, G.T.; Santos, R.A.; Touyz, R.M.; Burns, K.D. Angiotensin-(1-7) activates a tyrosine phosphatase and inhibits glucose-induced signalling in proximal tubular cells. Nephrol. Dial. Transplant. 2009, 24, 1766–1773.
- von Bohlen und Halbach, O.; Albrecht, D. The CNS renin-angiotensin system. Cell Tissue Res. 2006, 326, 599–616.
- Doobay, M.F.; Talman, L.S.; Obr, T.D.; Tian, X.; Davisson, R.L.; Lazartigues, E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R373–R381.
- Becker, L.K.; Etelvino, G.M.; Walther, T.; Santos, R.A.; Campagnole-Santos, M.J. Immunofluorescence localization of the receptor Mas in cardiovascular-related areas of the rat brain. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1416–H1424.
- Xue, B.; Zhang, Z.; Johnson, R.F.; Guo, F.; Hay, M.; Johnson, A.K. Central endogenous angiotensin-(1-7) protects against aldosterone/NaCl-induced hypertension in female rats. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H699–H705.
- Guimaraes, P.S.; Santiago, N.M.; Xavier, C.H.; Velloso, E.P.; Fontes, M.A.; Santos, R.A.; Campagnole-Santos, M.J. Chronic infusion of angiotensin-(1-7) into the lateral ventricle of the brain attenuates hypertension in DOCA-salt rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H393–H400.
- de Moura, M.M.; dos Santos, R.A.; Campagnole-Santos, M.J.; Todiras, M.; Bader, M.; Alenina, N.; Haibara, A.S. Altered cardiovascular reflexes responses in conscious Angiotensin-(1-7) receptor Mas-knockout mice. Peptides 2010, 31, 1934–1939.
- Heringer-Walther, S.; Batista, E.N.; Walther, T.; Khosla, M.C.; Santos, R.A.; Campagnole-Santos, M.J. Baroreflex improvement in shr after ace inhibition involves angiotensin-(1-7). Hypertension 2001, 37, 1309–1314.
- Britto, R.R.; Santos, R.A.; Fagundes-Moura, C.R.; Khosla, M.C.; Campagnole-Santos, M.J. Role of angiotensin-(1-7) in the modulation of the baroreflex in renovascular hypertensive rats. Hypertension 1997, 30, 549–556.
- Wu, Z.T.; Ren, C.Z.; Yang, Y.H.; Zhang, R.W.; Sun, J.C.; Wang, Y.K.; Su, D.F.; Wang, W.Z. The PI3K signaling-mediated nitric oxide contributes to cardiovascular effects of angiotensin-(1-7) in the nucleus tractus solitarii of rats. Nitric. Oxide 2016, 52, 56–65.
- Guimaraes, P.S.; Oliveira, M.F.; Braga, J.F.; Nadu, A.P.; Schreihofer, A.; Santos, R.A.S.; Campagnole-Santos, M.J. Increasing Angiotensin-(1-7) Levels in the Brain Attenuates Metabolic Syndrome-Related Risks in Fructose-Fed Rats. Hypertension 2014, 63, 1078–1085.
- Whitaker, A.M.; Molina, P.E. Angiotensin (1-7) contributes to nitric oxide tonic inhibition of vasopressin release during hemorrhagic shock in acute ethanol intoxicated rodents. Life Sci. 2013, 93, 623–629.
- Walther, T.; Balschun, D.; Voigt, J.P.; Fink, H.; Zuschratter, W.; Birchmeier, C.; Ganten, D.; Bader, M. Sustained long term potentiation and anxiety in mice lacking the Mas protooncogene. J. Biol. Chem. 2013, 273, 11867–11873.
- Almeida-Santos, A.F.; Kangussu, L.M.; Moreira, F.A.; Santos, R.A.; Aguiar, D.C.; Campagnole-Santos, M.J. Anxiolytic- and antidepressant-like effects of angiotensin-(1-7) in hypertensive transgenic (mRen2)27 rats. Clin. Sci. 2016, 130, 1247–1255.
- Braszko, J.J.; Karwowska-Polecka, W.; Halicka, D.; Gard, P.R. Captopril and Enalapril Improve Cognition and Depressed Mood in Hypertensive Patients. J. Basic Clin. Physiol. Pharmacol. 2003, 14, 323–343.
- Oscar, C.G.; de Figueiredo Muller-Ribeiro, F.C.; de Castro, L.G.; Lima, A.M.; Campagnolo-Santos, M.J.; Santos, R.A.S.; Xavier, C.H.; Fontes, M.A.P. Angiotensin-(1–7) in the basolateral amygdala attenuates the cardiovascular response evoked by acute emotional stress. Brain Res. 2015, 1594, 183–189.
- Souza, A.P.S.; Sobrinho, D.B.S.; Almeida, J.F.Q.; Alves, G.M.M.; Macedo, L.M.; Porto, J.E.; Vêncio, E.F.; Colugnati, D.B.; Santos, R.A.S.; Ferreira, A.J.; et al. Angiotensin II Type 1 receptor blockade restores angiotensin-(1-7)-induced coronary vasodilation in hypertrophic rat hearts. Clin. Sci. 2013, 125, 449–459.
- Costa, M.A.; Lopez Verrilli, M.A.; Gomez, K.A.; Nakagawa, P.; Peña, C.; Arranz, C.; Gironacci, M.M. Angiotensin-(1-7) upregulates cardiac nitric oxide synthase in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1205–H1211.
- Gomes, E.R.M.; Santos, R.A.S.; Guatimosim, S. Angiotensin-(1-7)-Mediated Signaling in Cardiomyocytes. Int. J. Hypertens. 2012, 2012.
- Ferreira, A.J.; Moraes, P.L.; Foureaux, G.; Andrade, A.B.; Santos, R.A.; Almeida, A.P. The angiotensin-(1-7)/Mas receptor axis is expressed in sinoatrial node cells of rats. J. Histochem. Cytochem. 2011, 59, 761–768.
- De Mello, W.C.; Ferrario, C.M.; Jessup, J.A. Beneficial versus harmful effects of Angiotensin (1-7) on impulse propagation and cardiac arrhythmias in the failing heart. J. Renin Angiotensin Aldosterone Syst. 2007, 8, 74–80.
- Lin, L.; Liu, X.; Xu, J.; Weng, L.; Ren, J.F.; Ge, J.; Zou, Y. Mas receptor mediates cardioprotection of angiotensin-(1-7) against Angiotensin II-induced cardiomyocyte autophagy and cardiac remodelling through inhibition of oxidative stress. J. Cell. Mol. Med. 2016, 20, 48–57.
- McCollum, L.T.; Gallagher, P.E.; Tallant, E.A. Angiotensin-(1-7) abrogates mitogen-stimulated proliferation of cardiac fibroblasts. Peptides 2012, 34, 380–388.
- Chang, R.L.; Lin, J.W.; Kuo, W.W.; Hsieh, D.J.; Yeh, Y.L.; Shen, C.Y.; Day, C.H.; Ho, T.J.; Viswanadha, V.P.; Huang, C.Y. Angiotensin-(1-7) attenuated long-term hypoxia-stimulated cardiomyocyte apoptosis by inhibiting HIF-1α nuclear translocation via Mas receptor regulation. Growth Factors 2016, 34, 11–18.
- Donoghue, M.; Wakimoto, H.; Maguire, C.T.; Acton, S.; Hales, P.; Stagliano, N.; Fairchild-Huntress, V.; Xu, J.; Lorenz, J.N.; Kadambi, V. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J. Mol. Cell. Cardiol. 2003, 35, 1043–1053.
- Wysocka, M.B.; Pietraszek-Gremplewicz, K.; Nowak, D. The Role of Apelin in Cardiovascular Diseases, Obesity and Cancer. Front. Physiol. 2018, 9, 557.
- Schiavone, M.T.; Santos, R.A.; Brosnihan, K.B.; Khosla, M.C.; Ferrario, C.M. Release of vasopressin from the rat hypothalamo-neurohypophysial system by angiotensin-(1-7) heptapeptide. Proc. Natl. Acad. Sci. USA 1988, 85, 4095–4098.
- Baracho, N.C.; Simões-e-Silva, A.C.; Khosla, M.C.; Santos, R.A. Effect of selective angiotensin antagonists on the antidiuresis produced by angiotensin-(1-7) in water-loaded rats. Braz. J. Med. Biol. Res. 1998, 31, 1221–1227.
- Magaldi, A.J.; Cesar, K.R.; de Araújo, M.; Simões e Silva, A.C.; Santos, R.A. Angiotensin-(1-7) stimulates water transport in rat inner medullary collecting duct: Evidence for involvement of vasopressin V2 receptors. Pflugers Arch. 2003, 447, 223–230.
- Ren, Y.; Garvin, J.L.; Carretero, O.A. Vasodilator action of angiotensin-(1-7) on isolated rabbit afferent arterioles. Hypertension 2002, 39, 799–802.
- van Twist, D.J.; Houben, A.J.; de Haan, M.W.; Mostard, G.J.; Kroon, A.A.; de Leeuw, P.W. Angiotensin-(1-7)-induced renal vasodilation in hypertensive humans is attenuated by low sodium intake and angiotensin II co-infusion. Hypertension 2013, 62, 789–793.
- Li, P.; Chappell, M.C.; Ferrario, C.M.; Brosnihan, K.B. Angiotensin-(1-7) augments bradykinin-induced vasodilation by competing with ACE and releasing nitric oxide. Hypertension 1997, 29, 394–400.
- Tallant, E.A.; Clark, M.A. Molecular mechanisms of inhibition of vascular growth by angiotensin-(1-7). Hypertension 2003, 42, 574–579.
- Fraga-Silva, R.A.; Costa-Fraga, F.P.; De Sousa, F.B.; Alenina, N.; Bader, M.; Sinisterra, R.D.; Santos, R.A. An orally active formulation of angiotensin-(1-7) produces an antithrombotic effect. Clinics 2011, 66, 837–841.
- Langeveld, B.; van Gilst, W.H.; Tio, R.A.; Zijlstra, F.; Roks, A.J. Angiotensin-(1-7) attenuates neointimal formation after stent implantation in the rat. Hypertension 2005, 45, 138–141.
- Sui, Y.B.; Chang, J.R.; Chen, W.J.; Zhao, L.; Zhang, B.H.; Yu, Y.R.; Tang, C.S.; Yin, X.H.; Qi, Y.F. Angiotensin-(1-7) inhibits vascular calcification in rats. Peptides 2013, 42, 25–34.
- Fang, C.; Stavrou, E.; Schmaier, A.A.; Grobe, N.; Morris, M.; Chen, A.; Nieman, M.T.; Adams, G.N.; LaRusch, G.; Zhou, Y.; et al. Angiotensin 1-7 and Mas decrease thrombosis in Bdkrb2-/- mice by increasing NO and prostacyclin to reduce platelet spreading and glycoprotein VI activation. Blood 2013, 121, 3023–3032.
- Sampaio, W.O.; Nascimento, A.A.S.; Santos, R.A.S. Regulation of Cardiovascular Signaling by Kinins and Products of Similar Converting Enzyme Systems. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1985–H1994.
- Sampaio, W.O.; Henrique de Castro, C.; Santos, R.A.S.; Schiffrin, E.L.; Touyz, R.M. Angiotensin-(1-7) counterregulates angiotensin II signaling in human endothelial cells. Hypertension 2007, 50, 1093–1098.
- Verano-Braga, T.; Schwämmle, V.; Sylvester, M.; Passos-Silva, D.G.; Peluso, A.A.B.; Etelvino, G.M.; Santos, R.A.S.; Roepstorff, P. Time-resolved quantitative phosphoproteomics: New insights into Angiotensin-(1-7) signaling networks in human endothelial cells. J. Proteome Res. 2012, 11, 3370–3381.
- Pernomian, L.; Gomes, M.S.; Restini, C.B.; de Oliveira, A.M. MAS-mediated antioxidant effects restore the functionality of angiotensin converting enzyme 2-angiotensin-(1-7)-MAS axis in diabetic rat carotid. BioMed Res. Int. 2014, 2014, 640329.
- Zhang, F.; Ren, J.; Chan, K.; Chen, H. Angiotensin-(1-7) regulates Angiotensin II-induced VCAM-1 expression on vascular endothelial cells. Biochem. Biophys. Res. Commun. 2013, 430, 642–646.
- Rentzsch, B.; Todiras, M.; Iliescu, R.; Popova, E.; Campos, L.A.; Oliveira, M.L.; Baltatu, O.C.; Santos, R.A.; Bader, M. Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension 2008, 52, 967–973.
- Alenina, N.; Xu, P.; Rentzsch, B.; Patkin, E.L.; Bader, M. Genetically altered animal models for Mas and angiotensin-(1-7). Exp. Physiol. 2008, 93, 528–537.
- Regenhardt, R.W.; Mecca, A.P.; Desland, F.; Ritucci-Chinni, P.F.; Ludin, J.A.; Greenstein, D.; Banuelos, C.; Bizon, J.L.; Reinhard, M.K.; Sumners, C. Centrally administered angiotensin-(1-7) increases the survival of stroke-prone spontaneously hypertensive rats. Exp. Physiol. 2014, 99, 442–453.
More