Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Caiyu Liu and Version 2 by Sirius Huang.
Metabolomics, as a new omics technology, has been widely accepted by researchers and has shown great potential in the field of nutrition and health. The process of metabolomics analysis includes sample preparation and extraction, derivatization, separation and detection, and data processing.
- metabolomics
- metabolites
1. Introduction
There are a variety of compounds in the metabolome, but there is still no good technology which can identify the components of these compounds fully and effectively at present. Metabolomics, as an important part of life science and biological system, has made significant contributions to the bioactive ingredients in food and their health effects on human beings through the analysis of changes in the metabolites [1]. Metabolomics is used to separate and identify small molecules in blood, urine, feces, cells, culture media, or food ingredients, and to study the related pathways [2]. The molecular weights of these small molecules are generally under 1000 Da [3].
The procedure of metabolomics analysis mainly involves the following steps: sample preparation, metabolite extraction, derivatization, separation and detection, and data processing (as shown in Figure 1) [3]. Small changes in any of these steps may have a significant impact on the final results.
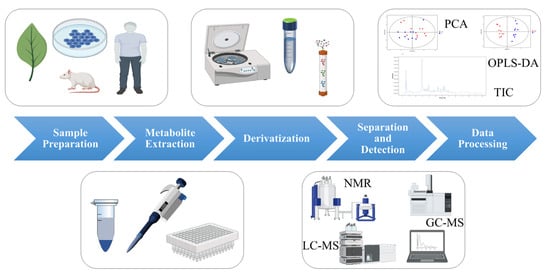
Figure 1. The process of metabolomics analysis. (Some picture elements are from the BioRender).
21. Sample Preparation
Sample preparation is the first step in metabolomics study. The quality of the prepared samples is the key to the success of metabolomics analysis, so it is very important to choose the appropriate preparation method for different samples.
The preparation methods are different for samples of various sources [4]. Keeping metabolite compositions of the original samples unchanged as much as possible and finding a suitable detection technology platform are two main problems in sample preparation. After the pretreatment of samples, choosing a suitable detection method can make the detection results have better repeatability and extraction efficiency [5]. For example, 4-chlorophenylalanine can be used to normalize the sample before GC-MS based metabolomics treatment to improve the extraction accuracy and efficiency [5]. Before the LC-MS based metabolomics sample is processed, the metabolites can be divided into different components by the mixed mode solid phase extraction method, and the appropriate column is selected to analyze the sample to improve the detection range of metabolites [6]. Methanol extraction can improve the quality of NMR spectra [7]. With the optimization of pretreatment technology, some special sample preparation methods have also been proposed. Solid phase microextraction (SPME) is widely accepted as a non-destructive method for the preparation of liquid samples [8]. Tijana Vasiljevic and colleagues proposed a method for preparing small samples of miniaturized SPME tips, which are coated with HLB particles [9]. It was the first study to analyze caviar samples using small SPME and LC successfully, and there was good extraction efficiency [9]. Wan Chan and colleagues compared the performance of several different serum preparation methods based on UPLC-MS, and found that serum samples prepared with methanol generated more accurate data [10]. In another study, it is showed that the speed of ultra-centrifugal treatment had a significant impact on the metabolic profile of fecal water; in particular, the concentration of P-Cresol changed with the increase of rotational speed [11]. However, this method is only suitable for NMR metabolomics studies at present.
For solid samples, freeze drying and grinding are required to reduce moisture in the samples and increase the release of metabolites, respectively. Quenching is a very important step to stop the metabolic processes, and this step includes adding liquid nitrogen, freezing, heating, and adding acid [12]. The omission of this step may cause changes in the metabolite composition by residual enzymes. However, time control is necessary in this step [12][13]. Sample preparation is a key step in metabolomics analysis [14]. How to prepare samples quickly without changing the original metabolite composition and make the operation repeatable are the problems to be solved in the future.
32. Metabolite Extraction
The step of metabolite extraction is generally the most rate-limiting step in metabolomics analysis [15]. There are different extraction methods for different types of samples to maximize the number, type, and concentration of the target metabolites. The selection of extraction solvent also has a significant effect on the recovery rate and metabolic profiles. The extraction solvents commonly used include water, chloroform, perchloric acid, methanol, acetonitrile, and other solutions [16][17]. It is necessary to choose hydrophilic solvents such as water-alcohol solutions for polar metabolites and hydrophobic solvents for non-polar metabolites. Estelle Martineau et al. compared the extraction efficiency of methanol/CHCl3/H2O, Acetonitrile/H2O, methanol/H2O, and Perchloric acid on mammalian cell metabolites, and found that using methanol/CHCl3/H2O for extraction can extract more metabolites, with good repeatability [15]. Karsten Seeger proposed a new method which extracts metabolites directly from NMR tubes by slice selection after centrifugation, and it provided a new idea for rapid determination of metabolites [18]. The most important thing of this method is that it could extract as many stable target metabolites as possible without adversely affecting subsequent analytical experiments [18].
43. Derivatization
This step is not always necessary. Generally, derivatization of the metabolites is required to transform the non-volatile compounds into volatile compounds to facilitate the analysis of metabolites and improve detection ability of the metabolites effectively if using GC-MS [19]. For example, the physicochemical properties of compounds with low ionization rate were changed by chemical derivatization to improve their ionization rate. Sezin Erarpat and colleagues used ultrasonic-assisted ethyl chloroformate to derivate l-methionine extract in human plasma and the recovery rate was up to 97.8 to 100.5% using GC-MS which could be regarded as a green and economical method [20]. Stable isotope labeling derivatization (SILD) is a novel sample pretreatment technology proposed in recent years, with a great potential in food metabolomics research based on LC-MS [21]. Shuyun Zhu et al. investigated a derivative method based on quadruplex stable isotope and developed 3-N-(D0-/D3-methyl-, and D0-/D5-ethyl-)-2’-carboxyl chloride rhodamine 6 G derivatization reagent, which can quickly and accurately quantify panaxadiol and panaxatriol in food [22]. Several studies have shown that derivatization can improve the ability of metabolite detection [23][24].
54. Separation and Detection
Separation and detection are important steps in metabolomics analysis. In the field of food nutrition, common separation technologies mainly include GC, LC, and capillary electrophoresis [25]. The separation of compounds is based on the adsorption capacity of each molecule in the stationary phase, and it is also related to the selection of column, eluent, fixation, and flow equivalence parameters [26]. In order to separate more metabolites, it is necessary to choose appropriate separation modes according to the polarity of the compounds. The separation technology is usually combined with high throughput detection technology to obtain large amounts of data. The commonly used detection techniques are NMR and MS [25]. Although the sensitivity of NMR is low, it can be used for non-invasive, rapid, and repeated analysis of a variety of metabolites at the μM levels [27]. It is simple to operate and suitable for high-throughput untargeted metabolomics analysis [28]. Both primary metabolites including amino acids, sugars, lipids, and organic acids, and secondary metabolites including flavonoids and alkaloids can be detected by NMR. By contrast, the sensitivity of MS is much higher, and it requires only a few μL of samples for analysis. MS can be combined with different separation techniques or in series according to different sample types [25]. GC-MS is mainly used to identify volatile and semi-volatile metabolites, while substances without volatile properties need to be derivatized, separated before detection by GC-MS. However, GC-MS cannot recognize any secondary metabolites [14]. Unlike GC-MS, LC-MS does not require complex pretreatment of samples, and it can directly separate and detect metabolites after extraction [14]. LC-MS is more comprehensive in metabolite identification and can determine secondary metabolites such as flavonoids as well as primary metabolites such as amino acids in plants.
Although current metabolomics techniques generally use a single detection tool, each technique has its own advantages and disadvantages. In order to identify and characterize more metabolites, combination of NMR and MS may achieve greater results. Manuja Kaluarachchi and colleagues identified metabolites in human plasma and serum by combination of 1H NMR and UPLC-MS [29]. They identified 4 metabolites with significant differences in plasma and serum by 1D NMR, and 10 other significant different metabolites by UPLC-MS, and most of them are found on glycerophospholipids [29]. Dong-sheng Zhao et al. determined the mechanisms of dioscorea bulbifera rhizome (DBR) on rat hepatotoxicity by integrating GC-MS and 1H NMR, and obtained a new potential therapeutic target, thus achieving an effective application of multi-platform metabolomics technology [30]. In addition, the introduction of chemicals in NMR tubes increased the likelihood of identifying compounds with specific physical and chemical properties; the 15N-edited NMR enabled specific binding to compounds containing free carbonyl [31]. The method of metabolic fingerprint analysis based on ultra-high-performance liquid chromatography–high-resolution mass spectrometry (UHPLC-HRMS) was optimized by using ethylene bridged hybrid C18 column, which showed good chromatographic resolution and realized the effective detection of infected metabolites in wheat [32]. Moreover, the optimization of parameters has been gradually studied. The researchers compared the Isotopologue Parameters Optimization (IPO) processing and manual processing of the original HPLC-TOF-MS data, and the parameters selected by IPO showed higher repeatability, and therefore it can be used to evaluate the optimum XCMS [33]. However, IPO need to take several days or even weeks to calculate the optimization parameters. In contrast, AutoTuner gives more robust and high-fidelity results [34]. MetaboAnalystR 3.0 is proposed as a new optimization process, which can not only optimize and correct parameters effectively, but also predict active pathways accurately [35]. In recent years, a hybrid metabolomics method based on mass spectrometry also attracted much attention. By bridging the advantages of targeted and untargeted metabolomics, more accurate results and more metabolites can be gained [36].
Different analytical instruments have different emphases. Considering the characteristics of samples and different analytical methods, a variety of separation and detection instruments can be used together to make the obtained metabolic data more comprehensive.
65. Data Processing
Data processing is an essential step in the process of metabolite screening, through which the changes of metabolites can be visualized and the possible metabolic pathways leading to these changes can be investigated using the KEGG database. Statistical analysis can help us to understand the metabolites in food and their impact on human health. Identification of metabolites is the most challenging step in metabolomics analysis [37]. The metabolites in the samples were obtained by comparing with the data in various resource databases. Choosing the right data processing method can greatly improve the accuracy of data analysis. There are many metabolome databases such as Metlin [29], Human Metabolome Database (HMDB) [38], KNApSack Database [39], and MassBank [40]. After aligning the information with these reliable databases and with multivariate statistical analysis, the obtained raw data can be converted to more meaningful conclusions, such as biomarkers.
Multivariate statistical analysis methods include principal component analysis (PCA), partial least squares discriminant analysis (PLS-DA), orthogonal partial least squares discriminant analysis (OPLS-DA), least absolute shrinkage and selection operator (LASSO), linear discriminant analysis (LDA), and so on [41][42]. Among which, PCA and PLS-DA are the most commonly used statistical methods in the field of metabolomics. PCA is a commonly used unsupervised dimensionality reduction method for metabolite quantity analysis, which reduces the data set to fewer dimensions to obtain greater variance [43]. It can help us to visualize the metabolic data, trend, and cluster. It has been reported recently that PCA was used in combination with quadrangular discriminating analysis (PCA-QDA) to identify the MS data of cancer samples, and its accuracy and specificity reached more than 90%, and therefore it can be called a satisfactory classification model [44]. PLS-DA is a supervised statistical analysis method that maximizes the correlation between variables, and it is often used to screen metabolites and to analyze overall metabolic changes between groups [45]. The availability of PLS-DA is good, and it can be used to process multiple dependent categorical variables simultaneously. However, PLS-DA is prone to overfitting [45]. In order to avoid this problem, based on the advantages of PLS-DA, OPLS-DA can divide the data into Y-related variation and Y-independent orthogonal variation and eliminate the variables unrelated to the experiment [46]. R2 and Q2 parameters are used to evaluate the prediction ability of the OPLS-DA model, and variable importance in the projection (VIP) can be generated from the model. VIP > 1.0 indicates that there are important potential biomarkers in the OPLS-DA model [47]. The authors compared PLS-DA with OPLS-DA in terms of model fitness and interpretability; although both are applicable, OPLS-DA had a higher interpretability [48]. At present, the co-analysis of PCA and OPLS-DA has become the mainstream trend of metabolomics to discriminate samples. Combination of more analytical models may be a future direction. Currently, the application of OPLS-DA is mainly to screen and identify biomarkers through s-plot/s-line, permutation, and VIP [49][50]. Using these methods to investigate the changes of metabolites may be a development direction in the future.
LASSO is a model selection method, and it can predict the phenotype by regression analysis of metabolites [51]. LDA can classify the samples according to the source and maximize the linear separation of the classes [52]. Kaitlyn M Mazzilli and colleagues evaluated the effects of various daily diets intake on serum metabolism using LASSO and found 102 related metabolites [53]. Virgilio Gavicho Uarrota et al. identified the metabolic components of cassava postharvest physiological deterioration (PPD) through PCA and PLS-DA models and realized good sample prediction [42]. The results provided good evidence for the metabolic differentiation of cassava during PPD. Moreover LDA and PCA in cluster analysis were considered to be suitable methods for distinguishing sex differences from organ differences [54]. For example, argininosuccinate showed significant differences between males and females in kidney tissue, and in the ventricle, males had significantly higher levels of free carnitine and total esterified carnitines than females [54]. So, targeted metabolomics is a good technique to test sex differences.
References
- Collins, C.; McNamara, A.E.; Brennan, L. Role of metabolomics in identification of biomarkers related to food intake. Proc. Nutr. Soc. 2019, 78, 189–196.
- Gonzalez-Dominguez, R.; Sayago, A.; Fernandez-Recamales, A. Metabolomics in Alzheimer’s disease: The need of complementary analytical platforms for the identification of biomarkers to unravel the underlying pathology. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1071, 75–92.
- Yang, Q.; Zhang, A.H.; Miao, J.H.; Sun, H.; Han, Y.; Yan, G.L.; Wu, F.F.; Wang, X.J. Metabolomics biotechnology, applications, and future trends: A systematic review. RSC Adv. 2019, 9, 37245–37257.
- Ulaszewska, M.M.; Weinert, C.H.; Trimigno, A.; Portmann, R.; Andres Lacueva, C.; Badertscher, R.; Brennan, L.; Brunius, C.; Bub, A.; Capozzi, F.; et al. Nutrimetabolomics: An Integrative Action for Metabolomic Analyses in Human Nutritional Studies. Mol. Nutr. Food Res. 2019, 63, e1800384.
- Yan, S.C.; Chen, Z.F.; Zhang, H.; Chen, Y.; Qi, Z.; Liu, G.; Cai, Z. Evaluation and optimization of sample pretreatment for GC/MS-based metabolomics in embryonic zebrafish. Talanta 2020, 207, 120260.
- Wu, Q.; Xu, Y.; Ji, H.; Wang, Y.; Zhang, Z.; Lu, H. Enhancing coverage in LC-MS-based untargeted metabolomics by a new sample preparation procedure using mixed-mode solid-phase extraction and two derivatizations. Anal. Bioanal. Chem. 2019, 411, 6189–6202.
- Yanibada, B.; Boudra, H.; Debrauwer, L.; Martin, C.; Morgavi, D.P.; Canlet, C. Evaluation of sample preparation methods for NMR-based metabolomics of cow milk. Heliyon 2018, 4, e00856.
- Reyes-Garcés, N.; Gionfriddo, E. Recent developments and applications of solid phase microextraction as a sample preparation approach for mass-spectrometry-based metabolomics and lipidomics. Trends Analyt. Chem. 2019, 113, 172–181.
- Vasiljevic, T.; Singh, V.; Pawliszyn, J. Miniaturized SPME tips directly coupled to mass spectrometry for targeted determination and untargeted profiling of small samples. Talanta 2019, 199, 689–697.
- Chan, W.; Zhao, Y.; Zhang, J. Evaluating the performance of sample preparation methods for ultra-performance liquid chromatography/mass spectrometry based serum metabonomics. Rapid Commun. Mass Spectrom. 2019, 33, 561–568.
- Yen, S.; Bolte, E.; Aucoin, M.; Allen-Vercoe, E. Metabonomic Evaluation of Fecal Water Preparation Methods: The Effects of Ultracentrifugation. Curr. Metab. 2018, 6, 57–63.
- Vuckovic, D. Sample preparation in global metabolomics of biological fluids and tissues. In Proteomic and Metabolomic Approaches to Biomarker Discovery; Academic Press: London, UK, 2020; pp. 53–83.
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221.
- Salem, M.A.; Perez de Souza, L.; Serag, A.; Fernie, A.R.; Farag, M.A.; Ezzat, S.M.; Alseekh, S. Metabolomics in the Context of Plant Natural Products Research: From Sample Preparation to Metabolite Analysis. Metabolites 2020, 10, 37.
- Martineau, E.; Tea, I.; Loaec, G.; Giraudeau, P.; Akoka, S. Strategy for choosing extraction procedures for NMR-based metabolomic analysis of mammalian cells. Anal. Bioanal. Chem. 2011, 401, 2133–2142.
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 2017, 19, 848–855.
- Kolho, K.L.; Pessia, A.; Jaakkola, T.; de Vos, W.M.; Velagapudi, V. Faecal and Serum Metabolomics in Paediatric Inflammatory Bowel Disease. J. Crohns Colitis 2017, 11, 321–334.
- Seeger, K. Simple and Rapid (Extraction) Protocol for NMR Metabolomics-Direct Measurement of Hydrophilic and Hydrophobic Metabolites Using Slice Selection. Anal. Chem. 2021, 93, 1451–1457.
- Karu, N.; Deng, L.; Slae, M.; Guo, A.C.; Sajed, T.; Huynh, H.; Wine, E.; Wishart, D.S. A review on human fecal metabolomics: Methods, applications and the human fecal metabolome database. Anal. Chim. Acta 2018, 1030, 1–24.
- Erarpat, S.; Bodur, S.; Ozturk Er, E.; Bakirdere, S. Combination of ultrasound-assisted ethyl chloroformate derivatization and switchable solvent liquid-phase microextraction for the sensitive determination of l-methionine in human plasma by GC-MS. J. Sep. Sci. 2020, 43, 1100–1106.
- Zhao, S.; Li, L. Dansylhydrazine Isotope Labeling LC-MS for Comprehensive Carboxylic Acid Submetabolome Profiling. Anal. Chem. 2018, 90, 13514–13522.
- Zhu, S.; Zheng, Z.; Peng, H.; Sun, J.; Zhao, X.E.; Liu, H. Quadruplex stable isotope derivatization strategy for the determination of panaxadiol and panaxatriol in foodstuffs and medicinal materials using ultra high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2020, 1616, 460794.
- Xue, G.; Su, S.; Yan, P.; Shang, J.; Wang, J.; Yan, C.; Li, J.; Wang, Q.; Xiong, X.; Xu, H. Integrative analyses of widely targeted metabolomic profiling and derivatization-based LC-MS/MS reveals metabolic changes of Zingiberis Rhizoma and its processed products. Food Chem. 2022, 389, 133068.
- Bian, X.; Li, N.; Tan, B.; Sun, B.; Guo, M.Q.; Huang, G.; Fu, L.; Hsiao, W.L.W.; Liu, L.; Wu, J.L. Polarity-Tuning Derivatization-LC-MS Approach for Probing Global Carboxyl-Containing Metabolites in Colorectal Cancer. Anal. Chem. 2018, 90, 11210–11215.
- Segers, K.; Declerck, S.; Mangelings, D.; Vander Heyden, Y.; Van Eeckhaut, A. Analytical techniques for metabolomic studies: A review. Bioanalysis 2019, 11, 2297–2318.
- Harrieder, E.M.; Kretschmer, F.; Bocker, S.; Witting, M. Current state-of-the-art of separation methods used in LC-MS based metabolomics and lipidomics. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2022, 1188, 123069.
- Wishart, D.S. NMR metabolomics: A look ahead. J. Magn. Reson. 2019, 306, 155–161.
- Agin, A.; Heintz, D.; Ruhland, E.; de la Barca, J.M.C.; Zumsteg, J.; Moal, V.; Gauehez, A.S.; Namer, I.J. Metabolomics—An overview. From basic principles to potential biomarkers (part 1). Med. Nucl.-Imag. Fonct. Metab. 2016, 40, 4–10.
- Kaluarachchi, M.; Boulange, C.L.; Karaman, I.; Lindon, J.C.; Ebbels, T.M.D.; Elliott, P.; Tracy, R.P.; Olson, N.C. A comparison of human serum and plasma metabolites using untargeted (1)H NMR spectroscopy and UPLC-MS. Metabolomics 2018, 14, 32.
- Zhao, D.S.; Wu, Z.T.; Li, Z.Q.; Wang, L.L.; Jiang, L.L.; Shi, W.; Li, P.; Li, H.J. Liver-specific metabolomics characterizes the hepatotoxicity of Dioscorea bulbifera rhizome in rats by integration of GC-MS and (1)H-NMR. J. Ethnopharmacol. 2018, 226, 111–119.
- Lane, A.N.; Arumugam, S.; Lorkiewicz, P.K.; Higashi, R.M.; Laulhe, S.; Nantz, M.H.; Moseley, H.N.; Fan, T.W. Chemoselective detection and discrimination of carbonyl-containing compounds in metabolite mixtures by 1H-detected 15N nuclear magnetic resonance. Magn. Reson. Chem. 2015, 53, 337–343.
- Rubert, J.; Righetti, L.; Stranska-Zachariasova, M.; Dzuman, Z.; Chrpova, J.; Dall’Asta, C.; Hajslova, J. Untargeted metabolomics based on ultra-high-performance liquid chromatography-high-resolution mass spectrometry merged with chemometrics: A new predictable tool for an early detection of mycotoxins. Food Chem. 2017, 224, 423–431.
- Alboniga, O.E.; Gonzalez, O.; Alonso, R.M.; Xu, Y.; Goodacre, R. Optimization of XCMS parameters for LC-MS metabolomics: An assessment of automated versus manual tuning and its effect on the final results. Metabolomics 2020, 16, 14.
- McLean, C.; Kujawinski, E.B. AutoTuner: High Fidelity and Robust Parameter Selection for Metabolomics Data Processing. Anal. Chem. 2020, 92, 5724–5732.
- Pang, Z.Q.; Chong, J.; Li, S.Z.; Xia, J.G. MetaboAnalystR 3.0: Toward an Optimized Workflow for Global Metabolomics. Metabolites 2020, 10, 186.
- Chen, L.; Zhong, F.Y.; Zhu, J.J. Bridging Targeted and Untargeted Mass Spectrometry-Based Metabolomics via Hybrid Approaches. Metabolites 2020, 10, 348.
- Dunn, W.B.; Erban, A.; Weber, R.J.M.; Creek, D.J.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J.; et al. Mass appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2013, 9, S44–S66.
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617.
- Afendi, F.M.; Okada, T.; Yamazaki, M.; Hirai-Morita, A.; Nakamura, Y.; Nakamura, K.; Ikeda, S.; Takahashi, H.; Altaf-Ul-Amin, M.; Darusman, L.K.; et al. KNApSAcK family databases: Integrated metabolite-plant species databases for multifaceted plant research. Plant Cell Physiol. 2012, 53, e1.
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714.
- Fang, H.; Zhang, A.H.; Sun, H.; Yu, J.B.; Wang, L.; Wang, X.J. High-throughput metabolomics screen coupled with multivariate statistical analysis identifies therapeutic targets in alcoholic liver disease rats using liquid chromatography-mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2019, 1109, 112–120.
- Uarrota, V.G.; Moresco, R.; Coelho, B.; Nunes Eda, C.; Peruch, L.A.; Neubert Ede, O.; Rocha, M.; Maraschin, M. Metabolomics combined with chemometric tools (PCA, HCA, PLS-DA and SVM) for screening cassava (Manihot esculenta Crantz) roots during postharvest physiological deterioration. Food Chem. 2014, 161, 67–78.
- Eicher, T.; Kinnebrew, G.; Patt, A.; Spencer, K.; Ying, K.; Ma, Q.; Machiraju, R.; Mathe, A.E.A. Metabolomics and Multi-Omics Integration: A Survey of Computational Methods and Resources. Metabolites 2020, 10, 202.
- Morais, C.; Lima, K. Principal Component Analysis with Linear and Quadratic Discriminant Analysis for Identification of Cancer Samples Based on Mass Spectrometry. J. Braz. Chem. Soc. 2017, 29, 472–481.
- Gromski, P.S.; Muhamadali, H.; Ellis, D.I.; Xu, Y.; Correa, E.; Turner, M.L.; Goodacre, R. A tutorial review: Metabolomics and partial least squares-discriminant analysis--a marriage of convenience or a shotgun wedding. Anal. Chim. Acta. 2015, 879, 10–23.
- Bujak, R.; Daghir-Wojtkowiak, E.; Kaliszan, R.; Markuszewski, M.J. PLS-Based and Regularization-Based Methods for the Selection of Relevant Variables in Non-targeted Metabolomics Data. Front. Mol. Biosci. 2016, 3, 35.
- Li, M.X.; Liu, M.Y.; Wang, B.Y.; Shi, L. Metabonomics Analysis of Stem Extracts from Dalbergia sissoo. Molecules 2022, 27, 1982.
- Homayoun, S.B.; Shrikant, I.B.; Kazem, M.; Hemmat, M.; Reza, M. Compared application of the new OPLS-DA statistical model versus partial least squares regression to manage large numbers of variables in an injury case-control study. Sci. Res. Essays 2011, 6, 4369–4377.
- Xue, W.; Zhang, H.; Liu, M.; Chen, X.; He, S.; Chu, Y. Metabolomics-based screening analysis of PPCPs in water pretreated with five different SPE columns. Anal. Methods 2021, 13, 4594–4603.
- Arendse, E.; Fawole, O.A.; Magwaza, L.S.; Nieuwoudt, H.; Opara, U.L. Evaluation of biochemical markers associated with the development of husk scald and the use of diffuse reflectance NIR spectroscopy to predict husk scald in pomegranate fruit. Sci. Hortic. 2018, 232, 240–249.
- Rohart, F.; Villa-Vialaneix, N.; Paris, A.; Canlet, C.; Sancristobal, M. Phenotypic Prediction Based on Metabolomic Data: LASSO vs. BOLASSO, Primary Data vs Wavelet Transformation. HAL Open Sci. 2010, 3–55. Available online: https://hal.archives-ouvertes.fr/hal-00658819 (accessed on 12 August 2022).
- Anowar, F.; Sadaoui, S.; Selim, B. Conceptual and empirical comparison of dimensionality reduction algorithms (PCA, KPCA, LDA, MDS, SVD, LLE, ISOMAP, LE, ICA, t-SNE). Comput. Sci. Rev. 2021, 40, 100378.
- Mazzilli, K.M.; McClain, K.M.; Lipworth, L.; Playdon, M.C.; Sampson, J.N.; Clish, C.B.; Gerszten, R.E.; Freedman, N.D.; Moore, S.C. Identification of 102 Correlations between Serum Metabolites and Habitual Diet in a Metabolomics Study of the Prostate, Lung, Colorectal, and Ovarian Cancer Trial. J. Nutr. 2020, 150, 694–703.
- Ruoppolo, M.; Caterino, M.; Albano, L.; Pecce, R.; Di Girolamo, M.G.; Crisci, D.; Costanzo, M.; Milella, L.; Franconi, F.; Campesi, I. Targeted metabolomic profiling in rat tissues reveals sex differences. Sci. Rep. 2018, 8, 4663.
More