Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Vivi Li and Version 1 by Arnoldo Aquino-Gálvez.
Hypoxia and hypoxia-inducible factors (HIFs) are essential in regulating several cellular processes, such as survival, differentiation, and the cell cycle; this adaptation is orchestrated in a complex way. The physiopathology of Idiopathic pulmonary fibrosis (IPF) has been proposed as epithelial-drive fibrosis, with converging genetic and environmental factors. Evidence has shown that these epithelial cell populations, particularly a group of basaloid cells identified by single-cell RNA sequencing (scRNA-seq) and the expression of marker senescence, development, and differentiation, are critical in the early stage of fibrotic lesions.
- lung regeneration
- IPF
- HIFs
- hypoxia
1. Introduction
Idiopathic pulmonary fibrosis (IPF) has a poor prognosis, with a median survival of 24–30 months [1[1][2],2], and is characterized by reduced functional capacity, dyspnea, and hypoxia induced by exercise or at rest [3,4,5][3][4][5]. Destruction of lung architecture impairs gas exchange and progresses to hypoxic respiratory failure, a hallmark of advanced disease [2]. The blood oxygen saturation level is considered an important parameter because its decrease during endurance tests predicts survival in patients with IPF [6]. Most patients have a poor quality of life due to low physical activity and limited exercise tolerance [2[2][7],7], decreased lung compliance leading to mechanical ventilation, and increased respiratory muscle energy expenditure, driving dyspnea [8,9][8][9].
Exertional dyspnea and worsening hypoxia are clinical features of IPF, and no drug is available to treat these two symptoms [10]; even oxygen inhalation does not improve tolerance to physical exertion in most patients and, therefore, does not relieve shortness of breath [11,12][11][12]. Obstructive sleep apnea (OSA) is known to be a risk factor for IPF; intermittent hypoxia (HI) and reoxygenation of OSA contribute to a poor prognosis [13]. Chronic exposure to HI increases mortality, lung inflammation, and pulmonary fibrosis in BLM-treated mice, suggesting a worse prognosis in patients who have IPF and severe OSA [13,14][13][14]. Most in vitro studies with cells and in vivo with animal models have shown that hypoxia is a determining factor in the progression and development of the disease. However, at the clinical level, there are still several questions.
The mechanisms by which hypoxia and transcription factors are involved have not been fully described. Initially, the relationship is directly proportional, since a higher degree of hypoxia is observed when the extracellular matrix accumulates. Notably, this effect was observed in fibroblasts, which are the cells in charge of the matrix remodeling; however, in light of recent discoveries in the pathophysiology of IPF, wresearchers suggest that the role of hypoxia has an impact beyond this.
IPF is an age-related and chronic lung disease characterized by alteration of the typical structure of the lung and progressive loss of respiratory capacity, whose etiology has not yet been elucidated [15]. IPF patients have few therapeutic options for antifibrotic drugs, which have shown limited efficacy, such as Pirfenidone and Nintedanib [16].
The physiopathology of IPF has been proposed as epithelial-drive fibrosis, with converging genetic and environmental factors [17]. At an early stage, lung injury is observed, with an aberrant response of epithelial cells that secrete many mediators for fibroblast migration and activation [18]. Recent evidence has shown that these epithelial cell populations, particularly a group of basaloid cells identified by single-cell RNA sequencing (scRNA-seq) and the expression of marker senescence, development, and differentiation, are critical in the early stage of fibrotic lesions [19]. These basaloid cells are lined with myofibroblasts activated in a complex microenvironment where hypoxia, through HIFs (HIF-1α, -2α, and -3α), could be involved in the establishment of profibrotic feedback promoting the development of IPF [20,21,22,23][20][21][22][23]. In addition, it has been shown that these mesenchymal cells can be helpful in the formation of the niche for alveolar regeneration [24,25][24][25]. Endothelial cells and their mediators are also involved in this interaction within alveoli [26]. Notably, Kobayashi et al. suggest that alveolar type-2 epithelial cells require a transitional state for terminal differentiation into type 1; this pre-alveolar type-1 transitional cell state (PATS) is a persistent phenomenon associated with failed regeneration in IPF [27]. Additionally, evidence shows that transdifferentiation of epithelial cells can be induced by interaction with aberrant mesenchymal cells [28]. Therefore, the role that hypoxia may have in differentiation and the possible regulation of epithelial cell transdifferentiation is essential to elucidate, since these cells are in constant interaction with cells that are in a hypoxic microenvironment.
2. Lung Oxygenation and Hypoxic Conditions
The lungs are responsible for capturing oxygen from the environment. In situations that lead to a decrease in oxygen tension, a pulmonary vascular response is activated to ensure adequate blood oxygenation [29]. In the lung, alveolar hypoxia causes vasoconstriction of the small pulmonary arteries. The mechanisms responsible for this vasoconstriction depend on pulmonary artery smooth muscle cells (PASMC) and endothelial cells, so the first sensor of oxygen depletion is the pulmonary vasculature [30,31][30][31]. In addition, pulmonary vasoconstriction due to hypoxia is mediated by increased intracellular calcium in human PASMC [32]. The availability of oxygen is crucial for cells to carry out different cellular processes, especially the production of energy in the form of adenosine triphosphate (ATP); the mitochondria are the central producer of ATP through metabolic pathways such as the tricarboxylic acid cycle (TCA) and oxidative phosphorylation (OXPHOS) [33,34][33][34]. When the demand for oxygen at the cellular level exceeds its supply, the cell enters a critical metabolic state that requires a strategic shift in its metabolism to adapt to low oxygen concentrations and maintain tissue survival (Figure 1) [35].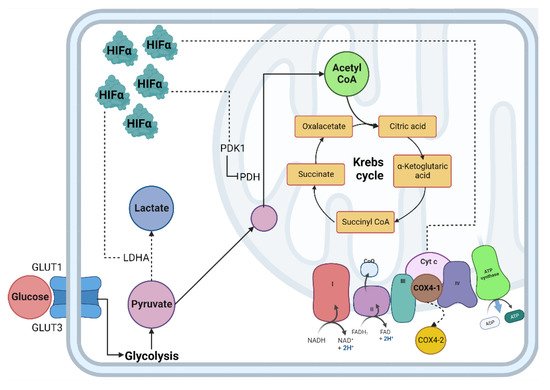
Figure 1. Cellular adaptation to hypoxia. The primary method of obtaining energy is the oxidation of glucose; under normoxic conditions, this begins in the cytosol through glycolysis, followed by the decarboxylation of pyruvate, then the TCA cycle where NADH and FADH2 are obtained, which, by giving up their electrons to the chain transport electrons, create an electrochemical proton gradient in the mitochondrial intermembrane and allow ATP synthase to release ATP molecules. In hypoxia, the enzymes involved in the glycolytic pathway, such as PDH, are inhibited by PDK1, decreasing the production of acetyl-CoA and increasing the production of lactate due to the activity of the enzyme LDH. Abbreviations: TCA: Tricarboxylic acid; NADH: reduced nicotinamide adenine dinucleotide; FADH2: reduced flavin adenine dinucleotide; ATP: adenosine-5’-triphosphate; PDH: pyruvate dehydrogenase; LDH: lactate dehydrogenase; HIF-α: Hypoxia Inducible Factor α subunit; HIF-β: Hypoxia Inducible Factor β subunit; Dotted line: HIF-α mitochondrial target gene.
3. Hypoxia-Inducible Factor-2α has a Particular Response in Idiopathic Pulmonary Fibrosis Pathogenesis
The pathological pattern of Usual Interstitial Pneumonia (UIP), characteristic of IPF, is heterogeneous, with areas of pulmonary fibrosis manifesting with foci of fibroblastic proliferation caused by epithelial damage and activation. These foci are located in the pulmonary interstitium and are characterized by the proliferation of fibroblasts and myofibroblasts, along with decreased apoptosis and hyperreactivity to fibrogenic cytokines [111][45]. Fibroblasts have a greater response capacity against profibrogenic cytokines, such as via transforming growth factor beta-1 (TGF-β1); this factor is involved in cellular functions such as cell proliferation, differentiation, and apoptosis [112][46], and the accumulation and activity of HIF-1α even under normoxic conditions, which in turn induces the expression of VEGF. This effect is enhanced by TGF-β1, which additionally inhibits the expression of the PHD2 gene through the Smad signaling pathway [113][47]. Fibroblasts constitute a diverse population of cells whose primary function is to establish, maintain, and modify the connective tissue stroma. In addition to interacting with various tissues, their primary function is to secrete proteins that constitute the extracellular matrix and play an essential role in wound repair, tissue development, and fibrosis [111][45]. In IPF, the population of pulmonary fibroblasts manifests a pathological phenotype, which may cause the perpetuation of this disease [114,115,116,117,118,119,120][48][49][50][51][52][53][54]. It has been suggested that activated fibroblasts acquire an aggressive phenotype and are the primary inductor of the accumulation of collagen in IPF [121][55]. Among the factors that induce this activation, wresearchers here highlight the role of TGF-β1 and hypoxia. Both induce the differentiation of fibroblasts to myofibroblasts, protection against apoptosis, and expression of α-actin in its cytoskeleton. In addition, this cytokine triggers the phenomenon known as Epithelial-Mesenchymal Transition (EMT) [122][56]. Notably, hypoxia promotes the proliferation in lung fibroblasts of patients with IPF, as well as in healthy subjects [123,124][57][58]. Significantly, hypoxia or another factor, such as TGF-β1, can promote the stabilization and activation of HIF-1α, favoring the production of LDH-5 not only in fibroblasts but also in the lung epithelium. These stimulate the differentiation of fibroblasts to myofibroblasts, forming a vicious cycle where hypoxia causes fibrosis and this, in turn, further promotes hypoxia [125][59]. HIFs are the main transcription factors that regulate the hypoxia response, so it is relevant to emphasize the structural differences of the HIF-1α and HIF-2α isoforms in specific proline residues, and the impact of those differences on the hypoxia response and IPF. An increasingly accepted hypothesis postulates that HIF-1α participates in regulating genes involved in the response to acute hypoxia, while HIF-2α has functions in chronic hypoxia [126,127][60][61]. In this context, hypoxia is acute when oxygen deprivation is for a short period, ranging from 2 to no more than 24 h, with an oxygen concentration of less than 1% [127,128][61][62]. The response to this type of hypoxia is mediated mainly by HIF-1α, which represents an initial response to hypoxia. In contrast, chronic hypoxia is hypoxia lasting more than 24 h, with an oxygen concentration of less than 5% [128][62]. In some studies, it has been observed that there is a change in the stabilization of HIF isoforms in neuroblasts and astrocytes during chronic exposure to a hypoxic environment, so this type of behavior supports the notion that the response to chronic hypoxic conditions is mainly mediated by HIF-2α [128,129][62][63]. HIF-1α has been reported to bind near the promoters, while HIF-2α binds to distal enhancers, and their distribution is not affected by the degree or duration of hypoxia or cell type. Additionally, the two isoforms do not compete for binding sites [130][64]. HIF-2α binds to distal enhancers, which could have functional repercussions that depend on other co-regulatory genes; for example, inhibition of HIF-2α function is produced by the recruitment of transcription cofactors or co-repressors in promoters of endogenous target genes [131][65]. WResearchers hypothesize that a delicate balance determines whether a tissue can activate the regeneration process or induce fibrosis, depending on the dysregulation and overexpression of HIFs. This work highlights the role of HIF-2α in the fibrogenic process. Recently, the dysregulated expression of HIF-2α has been proposed as a determining factor in the development of fibrosis; therefore, its overexpression in pulmonary fibroblasts from patients with IPF is probably the cause of the pseudohypoxia phenotype (or aerobic glycolysis) characteristic of these cells [22,132][22][66]. On the one hand, there is evidence that supports a correlation between hypoxia and the proliferation of IPF fibroblasts in a HIF-2α-dependent manner [21]. Genes that control cell proliferation and growth are regulated through HIF transcriptional factors [133][67]. Studies have shown that silencing of HIF-2α correlates with decreased miR-210 levels, leading to lower proliferation in fibroblasts derived from the lungs of IPF patients cultured in a hypoxic environment. In contrast, when HIF-1α is blocked, proliferation is not affected [21]. On the other hand, HIF-2α exclusively regulates some genes involved in VEGF activation in endothelial cells [52[68][69],134], correlating with the respiratory distress syndrome (RDS) observed in newborn mice caused by the loss of HIF-2α, which results in low production of surfactant by type II pneumocytes; the administration of VEGF prevents this syndrome [135][70]. Hypoxia increases VEGF-A expression in monocytes, fibroblasts, keratinocytes, myocytes, and endothelial cells [136,137,138,139][71][72][73][74]. This incremental increase in the production of VEGF-A in the different cell types that participate in regeneration and wound healing processes could be via activation of HIF-2α during hypoxia. In a previous study, wresearchers observed that the expression of α-smooth muscle actin (fibroblast to myofibroblast differentiation marker) resembles the expression pattern of HIF-2α, since both genes are overexpressed after 24 h of hypoxia exposure and are sustained for up to 96 h [22]. Recently, the HIF-1α signaling pathway has been identified as an essential factor in wound healing [140][75], and it is suggested that decreased regeneration could be related to changes in fibroblast activity [141][76]. HIF transcriptional activity has been reported to enable fibroblast to myofibroblast differentiation and the production of profibrotic mediators, where HIF signaling acts as an amplifier of IPF [56,113,142][47][77][78]. However, wresearchers consider that according to the evidence generated in animal models, regeneration depends on the regulation of oxygen by HIFs and, in turn, these promote a particular adaptation that affects the tissue repair process [109,110,143,144,145,146,147][79][80][81][82][83][84][85]. WResearchers recently proposed that hypoxia signaling pathways only make sense in the context of lung regeneration. Given that pulmonary fibrosis is assumed to be a product of damage and that the lung is trying to repair or regenerate itself, the hypoxia response would be necessary for the regeneration process; however, if it persists, it can lead to the activation of related feedback loops with disease progression [148][86]. More research is needed to elucidate the mechanisms involved in tissue regeneration. After tissue damage, a regeneration or scarring process can occur, but the determinants are still unclear; one of the reasons for this is probably due to evolutionary processes. Through animal regeneration models, HIFs have been determined to participate in these mechanisms. Evidence exists to reaffirm that fibrosis is an aberrant regeneration process that does not end. The kinetics of the regeneration process involves HIF-1α in the initial stage, HIF-2α in the intermediate stage, and HIF-3α in the final stage of regeneration. This does not happen in the same way in fibrosis, since multiple studies have indicated that in lung fibroblasts from patients with IPF and in other tissues with fibrosis, high levels of HIF-1α and HIF-2α are maintained (Figure 42). At the same time, HIF-3α is significantly decreased during hypoxia compared to healthy tissue or healthy lung fibroblasts in normoxia; therefore, wresearchers assume that this alteration does not allow a proper regeneration to be completed, and fibrosis persists.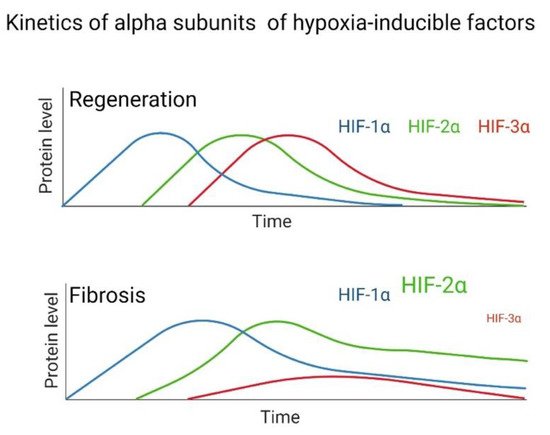
Figure 42.
Hypothesized difference in the kinetic pattern of HIFs between a fibrotic and regenerative model during hypoxia.
References
- Casanova, A.; Girón, R.M.; Molina, M.; Xaubet, A.; Ancochea, J. Predictive factors for survival in patients with idiopathic pulmonary fibrosis. Med. Clin. 2009, 133, 333–336.
- Krishna, R.; Chapman, K.; Ullah, S. Idiopathic Pulmonary Fibrosis; StatPearls Publishing: Treasure Island, FL, USA, 2022.
- Khor, Y.H.; Gutman, L.; Hussein, N.A.; Johannson, K.A.; Glaspole, I.N.; Guler, S.A.; Funke-Chambour, M. Incidence and Prognostic Significance of Hypoxemia in Fibrotic Interstitial Lung Disease: An International Cohort Study. Chest 2021, 160, 994–1005.
- Dowman, L.; Hill, C.J.; May, A.; Holland, A.E. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst. Rev. 2021, 2, CD006322.
- Andreas, G.; Krauss, E.; Tello, S.; Wagner, J.; Paul, B.; Kuhn, S.; Maurer, O.; Heinemann, S.; Costabel, U. The European IPF registry (eurIPFreg): Baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 141.
- Hallstrand, T.S.; Boitano, L.J.; Johnson, W.C.; Spada, C.A.; Hayes, J.G.; Raghu, G. The timed walk test as a measure of severity and survival in idiopathic pulmonary fibrosis. Eur. Respir. J. 2005, 25, 96–103.
- Chang, J.A.; Curtis, J.R.; Patrick, D.L.; Raghu, G. Assessment of health-related quality of life in patients with interstitial lung disease. Chest 1999, 116, 1175–1182.
- DiMarco, A.F.; Kelsen, S.G.; Cherniack, N.S.; Gothe, B. Occlusion pressure and breathing pattern in patients with interstitial lung disease. Am. Rev. Respir. Dis. 1983, 127, 425–430.
- Schaeffer, M.R.; Ryerson Christopher JRamsook, A.H.; Molgat-Seon, Y.; Wilkie, S.S.; Dhillon, S.S. Neurophysiological mechanisms of exertional dyspnoea in fibrotic interstitial lung disease. Eur. Respir. J. 2018, 51, 176.
- Geng, X.; Dufu, K.; Hutchaleelaha, A.; Xu, Q.; Li, Z.; Li, C.-M.; Patel, M.P.; Vlahakis, N.; Lehrer-Graiwer, J.; Oksenberg, D. Increased hemoglobin-oxygen affinity ameliorates bleomycin-induced hypoxemia and pulmonary fibrosis. Physiol. Rep. 2016, 4, e12965.
- Schaeffer, M.R.; Molgat-Seon, Y.; Ryerson, C.J.; Guenette, J.A. Supplemental oxygen and dypsnoea in interstitial lung disease: Absence of evidence is not evidence of absence. Eur. Respir. Rev. 2017, 26, 170033.
- Bell, E.C.; Cox, N.S.; Goh, N.; Glaspole, I.; Westall, G.P.; Watson, A. Oxygen therapy for interstitial lung disease: A systematic review. Eur. Respir. Rev. 2017, 26, 160080.
- Xiong, M.; Zhao, Y.; Mo, H.; Yang, H.; Yue, F.; Hu, K. Intermittent hypoxia increases ROS/HIF-1α ’related oxidative stress and inflammation and worsens bleomycin-induced pulmonary fibrosis in adult male C57BL/6J mice. Int. Immunopharmacol. 2021, 100, 108165.
- Gille, T.; Didier, M.; Rotenberg, C.; Delbrel, E.; Marchant, D.; Sutton, A. No TitleIntermittent Hypoxia Increases the Severity of Bleomycin-Induced Lung Injury in Mice. Oxid. Med. Cell. Longev. 2018, 2018, 1240192.
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961.
- Torrisi, S.E.; Kahn, N.; Vancheri, C.; Kreuter, M. Evolution and treatment of idiopathic pulmonary fibrosis. Presse Med. 2020, 49, 104025.
- Selman, M.; King, T.E.; Pardo, A.; American Thoracic Society; European Respiratory Society; American College of Chest Physicians. Idiopathic Pulmonary Fibrosis: Prevailing and Evolving Hypotheses about Its Pathogenesis and Implications for Therapy. Ann. Intern. Med. 2001, 134, 136.
- Selman, M.; Pardo, A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell. Signal. 2020, 66, 109482.
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983.
- Tzouvelekis, A.; Harokopos, V.; Paparountas, T.; Oikonomou, N.; Chatziioannou, A.; Vilaras, G.; Tsiambas, E.; Karameris, A.; Bouros, D.; Aidinis, V. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1α in disease pathogenesis. Am. J. Respir. Crit. Care Med. 2007, 176, 1108–1119.
- Bodempudi, V.; Hergert, P.; Smith, K.; Xia, H.; Herrera, J.; Peterson, M.; Khalil, W.; Kahm, J.; Bitterman, P.B.; Henke, C.A. miR-210 promotes IPF fibroblast proliferation in response to hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L283–L294.
- Aquino-Gálvez, A.; González-Ávila, G.; Jiménez-Sánchez, L.L.; Maldonado-Martínez, H.A.; Cisneros, J.; Toscano-Marquez, F.; Castillejos-López, M.; Torres-Espíndola, L.M.; Velázquez-Cruz, R.; Hugo, V.; et al. Dysregulated expression of hypoxia-inducible factors augments myofibroblasts differentiation in idiopathic pulmonary fibrosis. Respir. Res. 2019, 20, 130.
- Burman, A.; Blackwell, T.S.; Tanjore, H.; Burman, A.; Kropski, J.A.; Calvi, C.L.; Serezani, A.P.; Pascoalino, B.D.; Han, W.; Sherrill, T.; et al. Localized hypoxia links ER stress to lung fibrosis through induction of C/EBP homologous protein. JCI Insight 2018, 3, 16.
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255.
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123.
- Barratt, S.L.; Flower, V.A.; Pauling, J.D.; Millar, A.B. VEGF (Vascular endothelial growth factor) and fibrotic lung disease. Int. J. Mol. Sci. 2018, 19, 1269.
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 2020, 22, 934–946.
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.-D.; Wang, B.; Wolters, P.; et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5+ basal cells. Nat. Cell Biol. 2022, 24, 10–23.
- Urrutia, A.A.; Aragonés, J. HIF oxygen sensing pathways in lung biology. Biomedicines 2018, 6, 68.
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P.T. Hypoxic pulmonary vasoconstriction. Physiol Rev. 2012, 92, 367–520.
- Sommer, N.; Strielkov, I.; Pak, O.; Weissmann, N. Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur. Respir J. 2016, 47, 288–303.
- Yadav, V.R.; Song, T.; Mei, L.; Joseph, L.; Zheng, Y.M.; Wang, Y.X. PLCγ1-PKCε-IP3R1 signaling plays an important role in hypoxia-induced calcium response in pulmonary artery smooth muscle cells. Am. J. Physiol - Lung Cell Mol. Physiol. 2018, 314, L724–L735.
- Rich, P.R. The molecular machinery of Keilin’s respiratory chain. Biochem. Soc. Trans. 2003, 31, 1095–1105.
- Thomas, L.W.; Ashcroft, M. Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria. Cell Mol. Life Sci. 2019, 76, 1759–1777.
- Taylor, C.T. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem J. 2008, 409, 19–26.
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys Acta - Bioenerg 2010, 1797, 1171–1177.
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102.
- Hammond, E.M.; Asselin, M.C.; Forster, D.; O’Connor, J.P.B.; Senra, J.M.; Williams, K.J. The Meaning, Measurement and Modification of Hypoxia in the Laboratory and the Clinic. Clin. Oncol. 2014, 26, 277–288.
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours—Implications for treatment response. Br. J. Radiol. 2014, 87, 20130676.
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372.
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443.
- Dings, J.; Meixensberger, J.; Jäger, A.; Roosen, K. Clinical experience with 118 brain tissue oxygen partial pressure catheter probes: Comments. Neurosurgery 1998, 43, 1082–1094.
- Leach, R.M.; Treacher, D.F. Oxygen transport2. Tissue hypoxia. Bmj 1998, 317, 1370–1373.
- Müller, M.; Padberg, W.; Schindler, E.; Sticher, J.; Osmer, C.; Friemann, S.; Hempelmann, G. Renocortical tissue oxygen pressure measurements in patients undergoing living donor kidney transplantation. Anesth. Analg. 1998, 87, 474–476.
- Ancochea, J.; Antón, E.; Casanova, A. New therapeutic strategies in idiopathic pulmonary fibrosis. Arch. Bronconeumol. 2004, 40, 16–22.
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-β: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655.
- McMahon, S.; Charbonneau, M.; Grandmont, S.; Richard, D.E.; Dubois, C.M. Transforming growth factor β1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 2006, 281, 24171–24181.
- Negreros, M.; Hagood, J.S.; Espinoza, C.R.; Balderas-Martínez, Y.I.; Selman, M.; Pardo, A. Transforming growth factor beta 1 induces methylation changes in lung fibroblasts. PLoS ONE 2019, 14, e0223512.
- Im, J.; Nho, R.S. Fibroblasts from patients with Idiopathic Pulmonary Fibrosis are resistant to cisplatin-induced cell death via enhanced CK2-dependent XRCC1 activity. Apoptosis 2019, 24, 499–510.
- Jaffar, J.; Yang, S.H.; Kim, S.Y.; Kim, H.W.; Faiz, A.; Chrzanowski, W.; Burgess, J.K. Greater cellular stiffness in fibroblasts from patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L59–L65.
- Im, J.; Kim, K.; Hergert, P.; Nho, R.S. Idiopathic pulmonary fibrosis fibroblasts become resistant to Fas ligand-dependent apoptosis via the alteration of decoy receptor 3. J. Pathol. 2016, 240, 25–37.
- Habiel, D.M.; Hogaboam, C.M. Heterogeneity of Fibroblasts and Myofibroblasts in Pulmonary Fibrosis. Curr. Pathobiol. Rep. 2017, 5, 101–110.
- Luis-García, E.R.; Becerril, C.; Salgado-Aguayo, A.; Aparicio-Trejo, O.E.; Romero, Y.; Flores-Soto, E.; Mendoza-Milla, C.; Montaño, M.; Chagoya, V.; Pedraza-Chaverri, J.; et al. Mitochondrial Dysfunction and Alterations in Mitochondrial Permeability Transition Pore (mPTP) Contribute to Apoptosis Resistance in Idiopathic Pulmonary Fibrosis Fibroblasts. Int. J. Mol. Sci. 2021, 22, 7870.
- Waters, D.W.; Schuliga, M.; Pathinayake, P.S.; Wei, L.; Tan, H.Y.; Blokland, K.E.C.; Jaffar, J.; Westall, G.P.; Burgess, J.K.; Prêle, C.M.; et al. A senescence bystander effect in human lung fibroblasts. Biomedicines 2021, 9, 1162.
- Fernandez, I.E.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fi brosis in idiopathic pulmonary fi brosis. Lancet 2012, 380, 680–688.
- Powell, D.W.; Mifflin, R.C.; Valentich, J.D.; Crowe, S.E.; Saada, J.I.; West, A.B.; Myofibroblasts, I. Paracrine cells important in health and disease. Am. Physiol. Soc. 1998, 32, C1–C19.
- Mizuno, S.; Bogaard, H.J.; Voelkel, N.F.; Umeda, Y.; Kadowaki, M.; Ameshima, S.; Miyamori, I.; Ishizaki, T. Hypoxia regulates human lung fibroblast proliferation via p53-dependent and -independent pathways. Respir. Res. 2009, 10, 17.
- Senavirathna, L.K.; Huang, C.; Yang, X.; Munteanu, M.C.; Sathiaseelan, R.; Xu, D.; Henke, C.A.; Liu, L. Hypoxia induces pulmonary fibroblast proliferation through NFAT signaling. Sci. Rep. 2018, 8, 2709.
- Kottmann, R.M.; Kulkarni, A.A.; Smolnycki, K.A.; Lyda, E.; Dahanayake, T.; Salibi, R.; Honnons, S.; Jones, C.; Isern, N.G. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am. J. Respir. Crit. Care Med. 2012, 186, 740–751.
- Lin, Q.; Cong, X.; Yun, Z. Differential hypoxic regulation of hypoxia-inducible factors 1α and 2α. Mol. Cancer Res. 2011, 9, 757–765.
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423.
- Koh, M.Y.; Lemos, R., Jr.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1α- to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027.
- Guo, M.; Ma, X.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Zhao, Y. In chronic hypoxia, glucose availability and hypoxic severity dictate the balance between HIF-1 and HIF-2 in astrocytes. FASEB J. 2019, 33, 11123–11136.
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.E.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA-binding specificities of the HIF-1α and HIF-2α transcription factors in chromatin. EMBO Rep. 2019, 20, e46401.
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential Roles of Hypoxia-Inducible Factor 1α (HIF-1α) and HIF-2α in Hypoxic Gene Regulation. Mol. Cell. Biol. 2003, 23, 9361–9374.
- Brereton, C.J.; Yao, L.; Davies, E.R.; Zhou, Y.; Vukmirovic, M.; Bell, J.A.; Wang, S.; Ridley, R.A.; Dean, L.S.N.; Andriotis, O.G.; et al. Pseudohypoxic HIF pathway activation dysregulates collagen structure-function in human lung fibrosis. eLife 2022, 11, e69348.
- Huang, L.E.; Bunn, H.F. Hypoxia-inducible Factor and Its Biomedical Relevance. J. Biol. Chem. 2003, 278, 19575–19578.
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82.
- Elvert, G.; Kappel, A.; Heidenreich, R.; Englmeier, U.; Lanz, S.; Acker, T.; Rauter, M.; Plate, K.; Sieweke, M.; Breier, G.; et al. Cooperative interaction of hypoxia-inducible factor-2α (HIF-2α) and Ets-1 in the transcriptional activation of vascular endothelial growth factor receptor-2 (Flk-1). J. Biol. Chem. 2003, 278, 7520–7530.
- Compernolle, V.; Brusselmans, K.; Acker, T.; Hoet, P.; Tjwa, M.; Beck, H.; Plaisance, S.; Dor, Y.; Keshet, E.; Lupu, F.; et al. Loss of HIF-2α and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat. Med. 2002, 8, 702–710.
- Senger, D.R.; Ledbetter, S.R.; Claffey, K.P.; Papadopoulos-Sergiou, A.; Perruzzi, C.A.; Detmar, M. Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the α(v)β3 integrin, osteopontin, and thrombin. Am. J. Pathol. 1996, 149, 293–305.
- Möhle, R.; Green, D.; Moore, M.A.S.; Nachman, R.L.; Rafii, S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc. Natl. Acad. Sci. USA 1997, 94, 663–668.
- Brown, L.F.; Yeo, K.T.; Berse, B.; Yeo, T.K.; Senger, D.R.; Dvorak, H.F.; Van De Water, L. Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound Healing. J. Exp. Med. 1992, 176, 1375–1379.
- Frank, S.; Hubner, G.; Breier, G.; Longaker, M.T.; Greenhalgh, D.G.; Werner, S. Regulation of vascular endothelial growth factor expression in cultured keratinocytes. Implications for normal and impaired wound healing. J. Biol. Chem. 1995, 270, 12607–12613.
- Bonham, C.A.; Kuehlmann, B.; Gurtner, G.C. Impaired Neovascularization in Aging. Adv. Wound Care 2020, 9, 111–126.
- Vieira, W.A.; Wells, K.M.; McCusker, C.D. Advancements to the axolotl model for regeneration and aging. Gerontology 2016, 66, 212–222.
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019, 51, 1–13.
- hilip, K.; Mills, T.W.; Davies, J.; Chen, N.Y.; Karmouty-Quintana, H.; Luo, F.; Molina, J.G.; Amione-Guerra, J.; Sinha, N.; Guha, A.; et al. HIF1A up-regulates the ADORA2B receptor on alternatively activated macrophages and contributes to pulmonary fibrosis. FASEB J. 2017, 31, 4745–4758.
- Zhang, Y.; Strehin, I.; Bedelbaeva, K.; Gourevitch, D.; Clark, L.; Leferovich, J.; Messersmith, P.B.; Heber-Katz, E. Drug-induced regeneration in adult mice. Sci. Transl. Med. 2015, 7, 290ra92.
- Novianti, T.; Juniantito, V.; Jusuf, A.A.; Arida, E.A.; Jusman, S.W.A.; Sadikin, M. Expression and role of HIF-1α and HIF-2α in tissue regeneration: A study of hypoxia in house gecko tail regeneration. Organogenesis 2019, 15, 69–84.
- Simkin, J.; Gawriluk, T.R.; Gensel, J.C.; Seifert, A.W. Macrophages are necessary for epimorphic regeneration in African spiny mice. eLife 2017, 6, e24623.
- Zhang, Y.; Li, L.; Liu, Y.; Liu, Z.R. PKM2 released by neutrophils at wound site facilitates early wound healing by promoting angiogenesis. Wound Repair Regen. 2016, 24, 328–336.
- Sun, S.; Li, H.; Chen, J.; Qian, Q. Lactic acid: No longer an inert and end-product of glycolysis. Physiology 2017, 32, 453–463.
- Canhamero, T.; Garcia, L.V.; De Franco, M. Acute Inflammation Loci Are Involved in Wound Healing in the Mouse Ear Punch Model. Adv. Wound Care 2014, 3, 582–591.
- Santos, D.M.; Rita, A.M.; Casanellas, I.; Brito Ova, A.; Araújo, I.M.; Power, D.; Tiscornia, G. Ear wound regeneration in the African spiny mouse Acomys cahirinus. Regeneration 2016, 3, 52–61.
- Romero, Y.; Aquino-Gálvez, A. Hypoxia in cancer and fibrosis: Part of the problem and part of the solution. Int. J. Mol. Sci. 2021, 22, 8335.
More