Phosphatase and tensin homolog deleted on chromosome ten (PTEN) is a multifunctional tumor suppressor with protein- and lipid-phosphatase activities. The inactivation of PTEN is commonly found in all human cancers and is correlated with tumor progression. PTEN-lipid-phosphatase activity has been well documented to dephosphorylate phosphatidylinositol-3, 4, 5-phosphate (PIP3), which hinders cell growth and survival by dampening the PI3K and AKT signaling activity. PTEN-protein-phosphatase activity dephosphorylates the different proteins and acts in various cell functions.
- PTEN
- PTEN lipid phosphatase
- PTEN protein phosphatase
- mutation
- PTEN protein substrate
- tumorigenesis
1. PTEN Dual Lipid and Protein Phosphatase
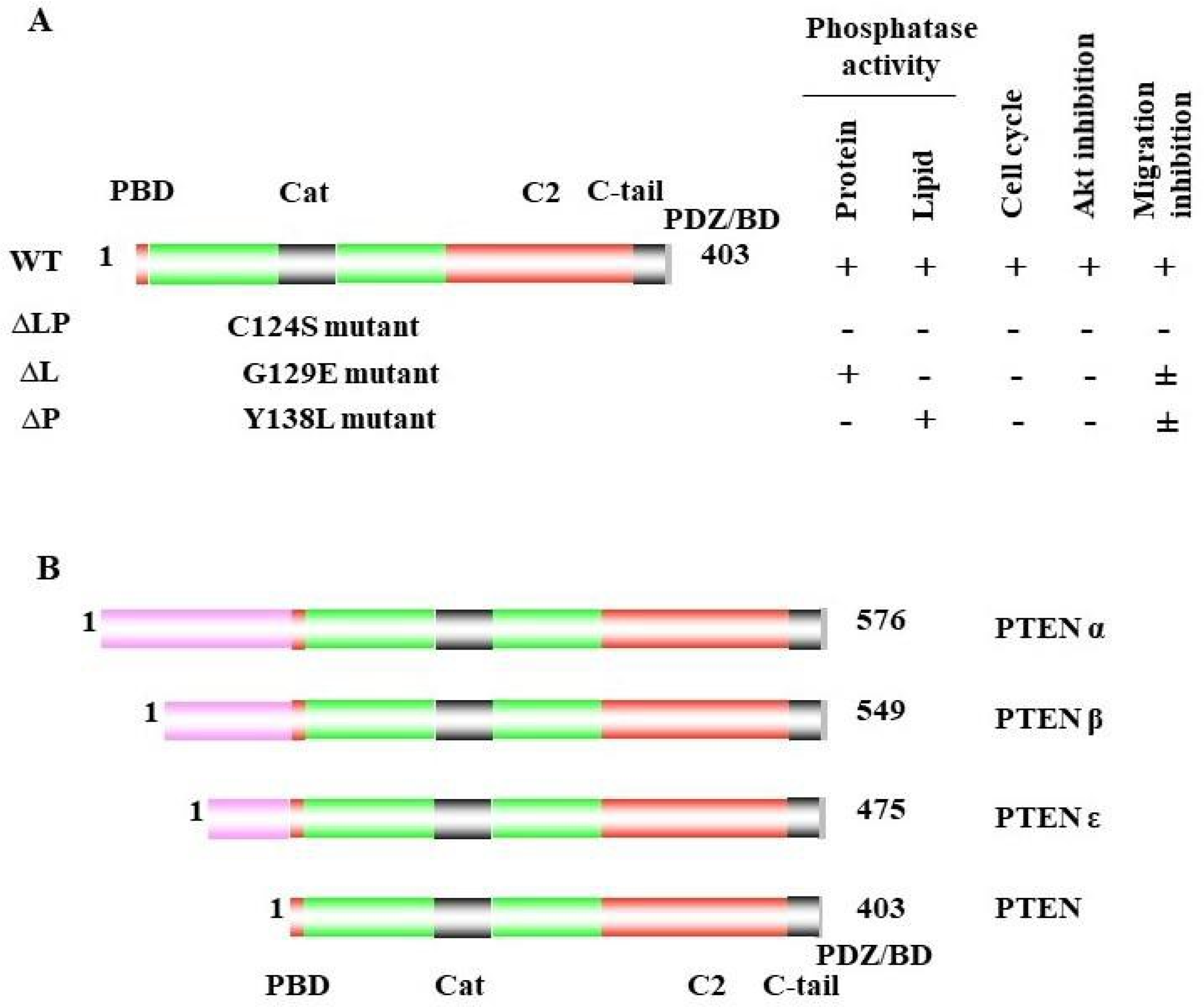
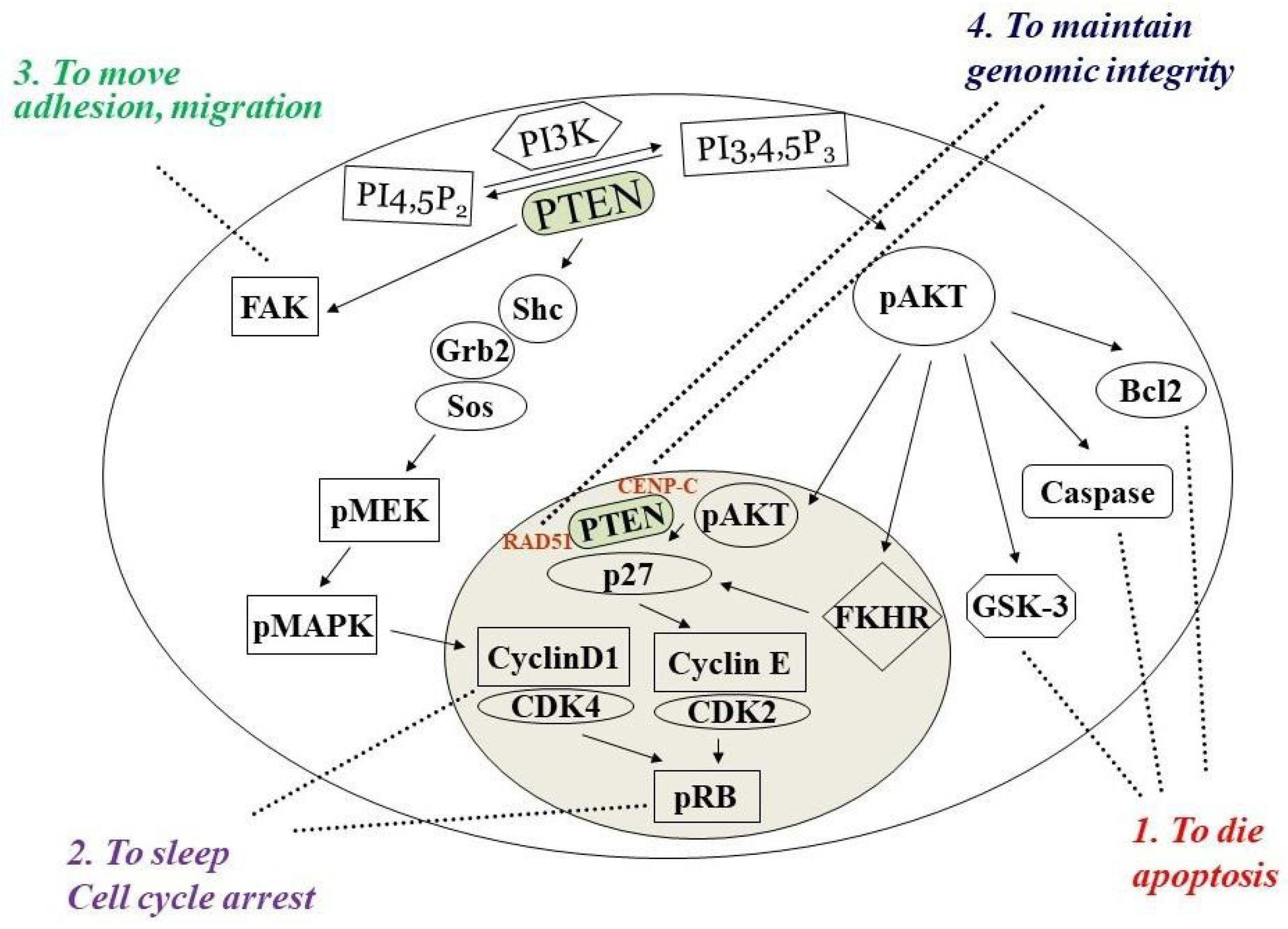
2. PTEN Inactivation in Cancer Progression
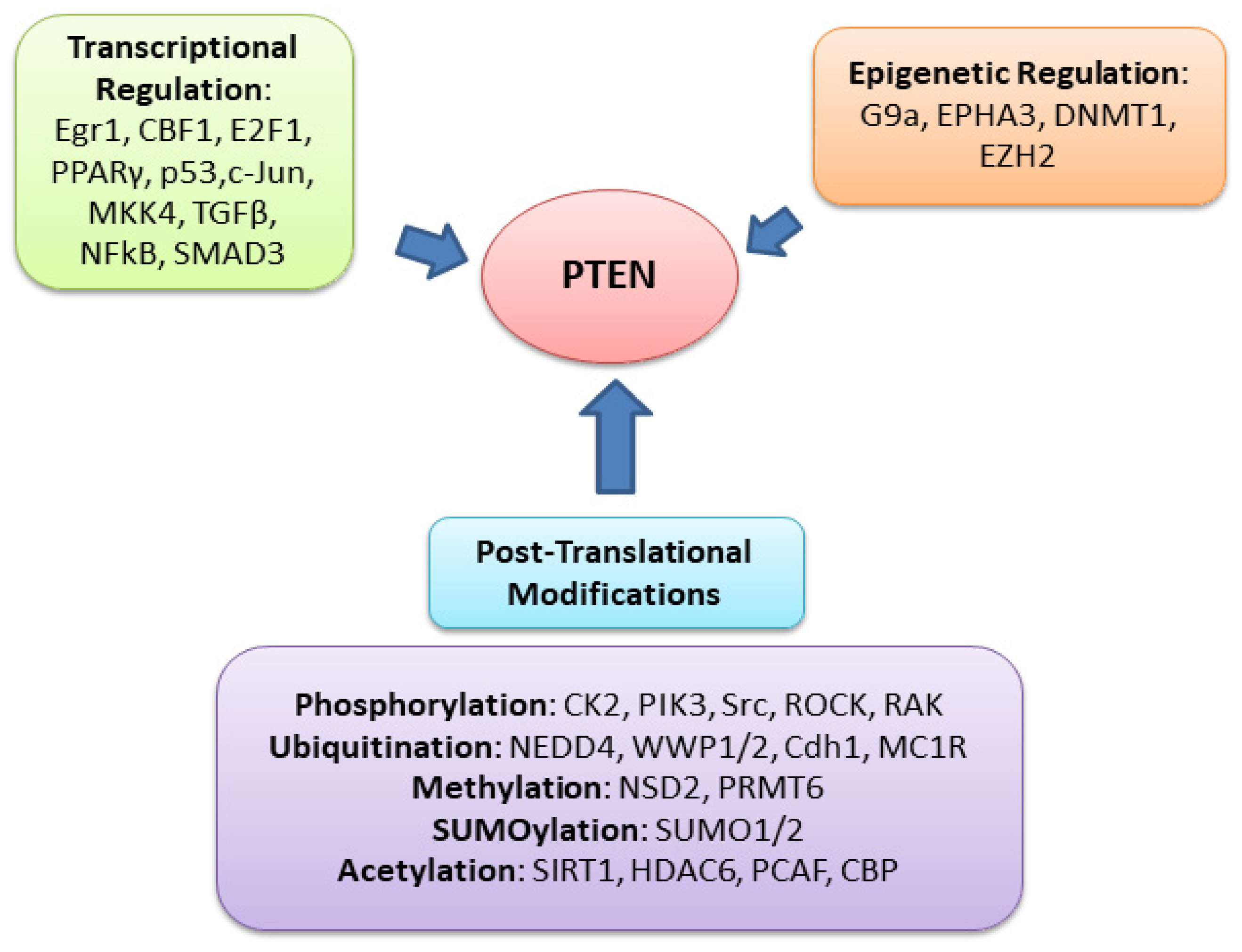
PTEN is one of the most highly mutated genes in human cancers. The dysfunction of PTEN by genetic mutation or epigenetic silencing contributes to most cancer development and progression (Figure 4, Table 1) [7,8,33][7][8][23]. It is often associated with high-grade and metastatic potential drug resistance [34,35][24][25] and poor patient prognosis [36,37,38][26][27][28]. The inactivation of PTEN is caused by various mechanisms, including genetic loss, point mutation, epigenetic regulation, and posttranslational modifications. Most alterations result in the loss of or reduction in PTEN protein.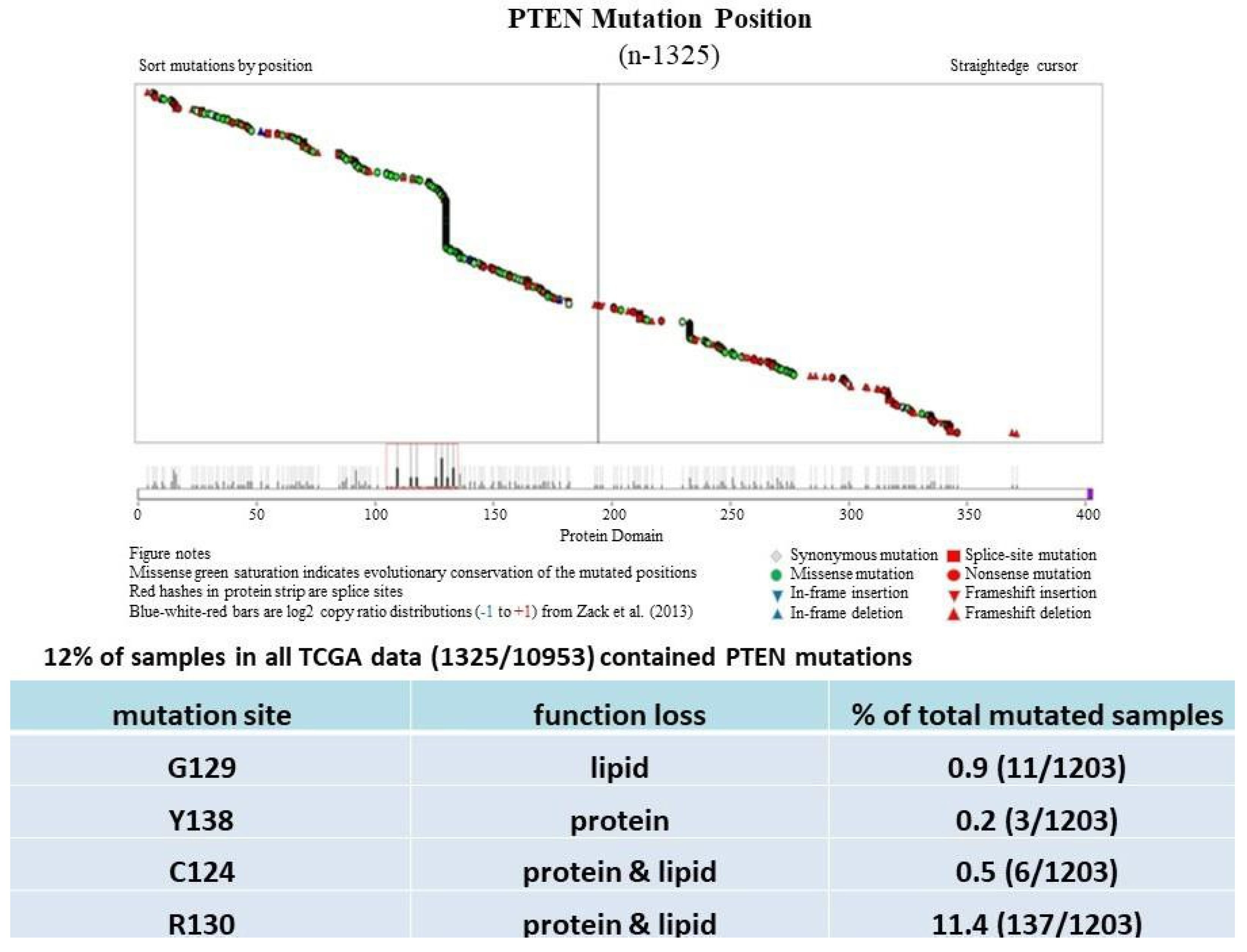
| Site/Tissue | Tumor Type | Range | Average | Comment | Reference(s) | |
|---|---|---|---|---|---|---|
| Brain | Glioblastoma | 12–84% | 29% (88/303) | Mostly LOH | [53,54] | [29][30] |
| Breast | Ductal carcinoma | 11–55% | 33% (415/1257) | Mostly LOH | [55,56,57] | [31][32][33] |
| Endometrium | Endometrioid carcinoma | 19–82% | 67% (352/529) | LOH and mutation | [58,59] | [34][35] |
| Prostate | Adenocarcinoma | 12–63% | 33% (88/267) | Mostly LOH | [60,61] | [36][37] |
| Ovary | Cystadenocarcinoma | 9–61% | 30% (33/112) | LOH and mutation | [62,63] | [38][39] |
| Skin | Melanoma | 11–39% | 30% (57/190) | Mostly LOH | [64,65] | [40][41] |
| Thyroid | Carcinoma (ATC) | 10–41% | 11% (21/196) | Mostly LOH | [66,67,68] | [42][43][44] |
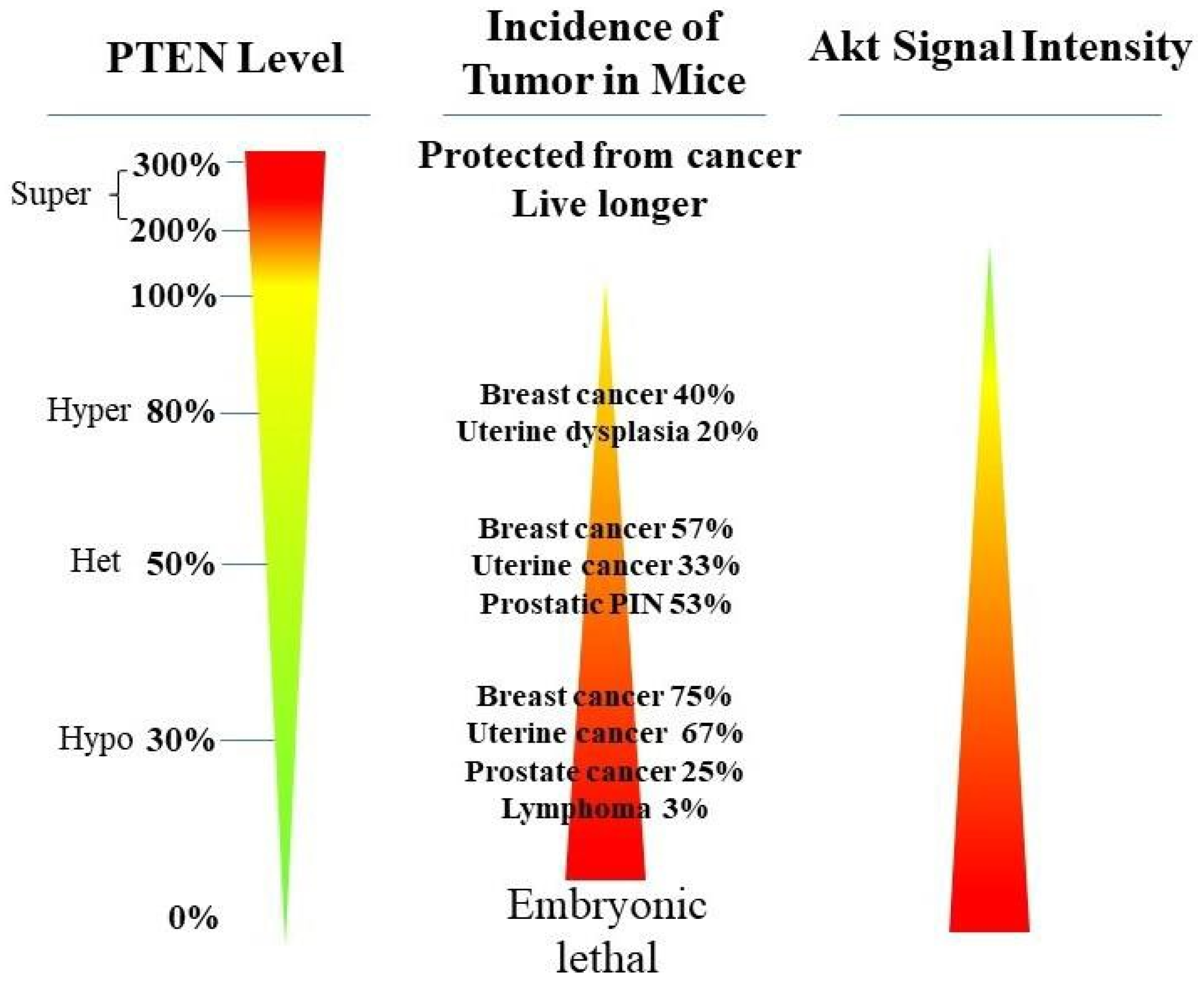
PTEN genetic loss is involved in various tumor types, such as glioblastoma, breast ductal carcinoma, endometrial carcinoma, prostate adenocarcinoma, ovary cystadenocarcinoma, melanoma, pancreatic adenocarcinoma, colorectal cancer, etc. (Table 1). In GEM mice, PTEN deficiency leads to embryonic lethality, and even a small reduction in the PTEN levels enhances the cancer incidence [23,24,39,40][20][21][45][46]. Contrarily, the systemic overexpression of PTEN in GEM models amplifies its tumor-suppressive function and protects against tumorigenesis [41][47], which suggests that the precise level of PTEN expression is a critical factor for the tumor-suppressor function, and that the reduction in PTEN activity is a driving mechanism for tumor progression. The pathologic characteristics that are associated with PTEN deletion in mice are phenocopied in a wide range of human tumors with loss-of-heterozygosity (LOH) mutations. In cancer, the PI3K/PTEN/Akt pathway has been identified as one of the critical molecular axes driving tumorigenesis [42,43,44,45][48][49][50][51]. The loss of PTEN activity has been reported to be responsible for many of the phenotypes of cancers, and it affects the development of 15–70% of human cancers [35,43,44][25][49][50] (Table 1).
Much has been learned about the features of PTEN point mutation through the analysis of PTEN germline mutations found in patients with Cowden disease (CD) [46][52]. Patients with CD, who harbor missense mutations in the PTEN phosphatase domain, are cancer-prone and develop more lesions than patients with PTEN deletion, which causes the complete loss of the PTEN function [47][53].
4. PTEN-Protein-Phosphatase Substrates and PI3K-Independent Function
4.1. PTEN
PTEN has been reported to play a vital role in regulating cell migration, invasion, and metastasis [48,115,116,117,118][69][72][73][74][75]. As indicated above, the naturally derived PTEN point mutant G129E [9[9][75][76],118,119], which loses lipid but maintains protein-phosphatase activity, retains the ability to inhibit cell migration and invasion, as well as metastasis [48][69]. To study the other mechanisms involved in cell migration, Reftopoulou and colleagues found that PTEN protein phosphatase is required for migration. They further demonstrated that PTEN protein phosphatase dephosphorylates its C domain at Thr383, inhibiting the migration independent of its effects on the PI3K pathway [21][61].4.2. Abi1
Abl-interactor 1 (Abi1) is a core component of the WASP-family verprolin homologous protein (WAVE) regulatory complex (WRC). Abi1 acts as a core scaffold protein to mediate the membrane recruitment and stabilization of WRC subunits [122[77][78],123], and it is regulated by extracellular cues and intracellular signaling pathways. PTEN can bind and dephosphorylate Abi1at Y213 and S216, triggering its degradation through the calpain pathway, thereby promoting epithelial differentiation and polarization [99][79], as well as epithelial–mesenchymal transition and cancer-stem-cell activity [124][80]. PTEN dephosphorylates Abi1 and downregulates the WRC at the cell cortex, thereby reorganizing the actin cytoskeleton to facilitate the formation of the apical actin belt and adherent junctions.4.3. Β-Catenin
Transforming growth factor β (TGF-β) is a pleiotropic cytokine that plays a role in growth suppression in normal epithelial cells, but it supports metastasis formation in many tumors [125][81].4.4. Cofilin-1
Cofilin-1 is an essential actin regulator. As an actin-depolymerizing factor (ADF)/cofilins family protein, Cofilin-1 regulates the rapid depolymerization of actin microfilaments that give actin its characteristic dynamic instability and its central role in muscle contraction, cell motility, and transcription regulation [126,127,128][82][83][84]. The activity of cofilin is regulated by a variety of mechanisms, including specific phosphorylation and dephosphorylation [128][84].4.5. CREB
The transcription factor cyclic AMP response element-binding protein (CREB) is activated via phosphorylation at serine133, which mediates gene transcription and promotes cell proliferation and survival. PTEN protein phosphatase is required for the dephosphorylation of CREB in the nucleus. Under PTEN-deficient conditions, CREB phosphorylation is enhanced independently of the PI3K/AKT pathway. The inhibition of the PI3K/AKT pathway does not affect the CREB phosphorylation in PTEN-deficient cells. C124S-mutant PTEN cannot dephosphorylate CREB, while G129E and wild-type PTEN can, which demonstrates that the protein-phosphatase activity of PTEN is essential for dephosphorylating and colocalizing with CREB in the nucleus [102][85], which suggests that PTEN regulates gene expression through this mechanism.4.6. Drebrin
Drebrin is a protein that is encoded by the DBN1 gene. It is a crucial regulator of the actin cytoskeleton in neuronal cells for synaptic plasticity, neurogenesis, and neuronal migration, as well as in cancer cells for tumor invasion [132,133,134][86][87][88]. The defect of Drebrin in expression and activation contributes to the pathogenesis.4.7. Dvl
PTEN is an essential regulator of multicilia formation and cilia disassembly via Dishevelled (DVL2) phosphorylation. DVL is an important component of the WNT signaling pathways that plays a role during convergent extension movements. DVL is a ciliogenesis regulator in Xenopus and human epithelial cells, and it has been identified as a direct substrate for PTEN. Among the DVL proteins, DVL2 and DVL3 have the strongest associations with PTEN. The knockdown of PTEN increases the DVL2 phosphorylation on serine143 during cilia formation. By studying wild-type PTEN and mutants of PTEN (C124S, G129E, Y138L), it was confirmed that the protein-phosphatase activity of PTEN is responsible for directly dephosphorylating DVL2 on serine 143 [104][89], which implicates PTEN in multicilia formation and movement.4.8. FAK
Focal adhesion kinase (FAK) is one of the earliest confirmed substrates for PTEN. PTEN overexpression inhibits cell migration via reducing the phosphorylation of FAK. The effect of PTEN on cell spreading was examined on fibronectin (FN) in multiple cell lines: NIH 3T3 cells, human fibroblast cells, DBTRG-05MG cells (glioblastoma), and U-87MG (glioma) cells. The overexpression of PTEN delayed or inhibited the spreading in these cell lines [48][69]. FAK was first found to be a substrate of the Src proto-oncogene. It is an important regulator of cell adhesion and motility. The tyrosine phosphorylation of FAK is associated with focal contacts that form at ECM integrin junctions, and integrin-binding proteins recruit FAK to the focal contacts. It has been reported that PTEN suppresses cell migration, invasion, and metastasis through the dephosphorylation of FAK at Tyr397 [48,105][69][90]; however, specific FAK dephosphorylation sites have not been observed in other cell types [136][91], and other mechanistic details have yet to be uncovered.4.9. Glucocorticoid Receptor (GR)
GR is a pleiotropic nuclear receptor and transcriptional regulatory factor that controls the network of glucocorticoid (GC)-responsive genes in a positive or negative manner for regulating numerous physiological and cellular processes [137][92]. In cancer, GR activation also appeared as tumor-suppressing [138][93] and tumor-promoting effects [139][94].
4.10. IRS1
PTEN protein phosphatase can dephosphorylate insulin receptor substrate-1 (IRS1). IRS1 is a mediator of insulin and IGF signaling, which is negatively affected by NEDD4. In NEDD4-deficient cells, IGF signaling becomes defective, while AKT activation is unimpaired. PTEN ablation rescues impaired IGF signaling by dephosphorylating IRS1. Although NEDD4 is required for IGF signaling, the role of NEDD4 in IGF signaling is PTEN-dependent. NEDD4 inhibits the PTEN function to enable IGF signaling. NEDD4 is responsible for the ubiquitination of PTEN, and for suppressing its phosphatase activity. The C124S mutant of PTEN is not able to dephosphorylate IRS1, while the G129E mutant and wild-type PTEN are, which proves that IRS1 is a direct substrate of PTEN’s protein phosphatase [106][95].4.11. MCM2
The MCM2-7 complex is one of the core components of the replisome [140[96][97],141], and it plays crucial roles in replication origin firing, elongation, termination, and the replication-stress response [142,143][98][99]. MCM2 is a critical component of the MCM2-7 complex, and it is a core replication helicase of the replisome that is regulated by the MCM2 phosphorylation status.4.12. NKX3.1
NKX3.1, which is a prostate-specific homeobox gene, is a gatekeeper suppressor and is commonly deleted in prostate cancer. NKX3.1 inhibits cell proliferation and mediates cell apoptosis and DNA repair.4.13. PLK1
Polo-like kinase 1 (PLK1) is a mitotic kinase that regulates mitotic entry and exit. PLK1 controls spindle bipolarity and is involved in cytokinesis. PTEN physically associates with PLK1 and dephosphorylates PLK1, maintaining genomic stability during cell division. PTEN loss or deficiency causes failure in cytokinesis through nondisjunction chromosomes and cleavage-furrow regression, which leads to spontaneous polyploidy and resistance to spindle disruption.4.14. PTK6
PTEN inhibits protein tyrosine kinase 6 (PTK6/BRK/Sik) activity in prostate cancer cells by dephosphorylating PTK6 at tyrosine 342 (PY342). In the absence of PTEN, PTK6 is activated and downstream oncogenic signaling is promoted [111][100].4.15. Pol II
The RNA polymerase II (Pol II) C-terminal domain (CTD) constantly undergoes cycles of phosphorylation/dephosphorylation during gene transcription. It was demonstrated that PTEN dephosphorylates Pol II CTD with specificity for Ser5, and that the phosphorylation of Pol II CTD Ser5 is inversely related to PTEN expression. When there is an overexpression of PTEN, there will be a decrease in the Pol II CTD phosphorylation; global Pol II CTD phosphorylation increases along with the PTEN loss. Pol II CTD is a significant platform for posttranslational modifications, such as elongation, termination, and co-transcriptional processes. PTEN, as a Pol II CTD phosphatase, raises the possibility that PTEN can regulate global transcription [110][101].4.16. Rab7
Rab7 is a GTPase for endosome maturation that is involved in epidermal growth factor receptor (EGFR) signaling. PTEN dephosphorylates Rab7 on two residues, S72 and Y183, and it promotes late endosome maturation, which reduces EGFR signaling. The residues are required to associate Rab7 with GDP dissociation inhibitor (GDI)-dependent recruitment to late endosomes and maturation. EGFR is a receptor tyrosine kinase that regulates cell proliferation, growth, and motility. PTEN controls the EGFR-endocytic-trafficking pathway via the dephosphorylation of Rab7 and the localization of Rab7. The loss of PTEN causes EGFR transport from early to late endosomes because PTEN is needed for Rab7-endosomal-membrane targeting. PTEN-mediated Rab7 dephosphorylation allows Rab7 to interact with GDI, GEF, and effector proteins. Rab7 has been identified as a protein substrate for PTEN, which provides a new mechanism for controlling the EGFR signaling in cells via PTEN [112][102].4.17. Shc
Src homology collagen (Shc) is a direct substrate for PTEN protein phosphatase. Shc is an SH2-binding adaptor molecule that activates the Raf and MAPK pathway. PTEN inhibits the MAPK pathway by dephosphorylating Shc in positions Tyr239/240, which reduces cell proliferation and inhibits Shc-mediated tumor metastasis in renal-cell carcinoma (RCC) [113][103].4.18. SRC
SRC is a membrane-anchored tyrosine kinase that is activated following the engagement of many different classes of cellular receptors, and it regulates various biological activities, including cell proliferation, adhesion, migration, and transformation [144][104]. The study reported that SRC modulates the antibody Trastuzumab that targets the human epithermal growth factor receptor-2 (HER-2 or ERBB2) response in breast cancer, and that activating SRC by phosphorylation at Tyr416 was required for regulating the multiple resistance pathways [114][105].References
- Bigner, S.H.; Mark, J.; Mahaley, M.S.; Bigner, D.D. Patterns of the early, gross chromosomal changes in malignant human gliomas. Hereditas 1984, 101, 103–113.
- Gibas, Z.; Prout, G.R., Jr.; Connolly, J.G.; Pontes, J.E.; Sandberg, A.A. Nonrandom chromosomal changes in transitional cell carcinoma of the bladder. Cancer Res. 1984, 44, 1257–1264.
- Lundgren, R.; Kristoffersson, U.; Heim, S.; Mandahl, N.; Mitelman, F. Multiple structural chromosome rearrangements, including del(7q) and del(10q), in an adenocarcinoma of the prostate. Cancer Genet. Cytogenet. 1988, 35, 103–108.
- Hsu, S.C.; Volpert, O.V.; Steck, P.A.; Mikkelsen, T.; Polverini, P.J.; Rao, S.; Chou, P.; Bouck, N.P. Inhibition of an-gio-genesis in human glioblastomas by chromosome 10 induction of thrombospondin-1. Cancer Res. 1996, 56, 5684–5691.
- Ittmann, M. Allelic loss on chromosome 10 in prostate adenocarcinoma. Cancer Res. 1996, 56, 2143–2147.
- Li, D.M.; Sun, H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997, 57, 2124–2129.
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science 1997, 275, 1943–1947.
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.A.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362.
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. 607 Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67.
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334.
- Kotelevets, L.; Trifault, B.; Chastre, E.; Scott, M.G. Posttranslational Regulation and Conformational Plasticity of PTEN. Cold Spring Harb. Perspect. Med. 2020, 10, a036095.
- Pulido, R. PTEN: A yin-yang master regulator protein in health and disease. Methods 2015, 77, 3–10.
- Iwasaki, H.; Murata, Y.; Kim, Y.; Hossain, I.; Worby, C.A.; Dixon, J.E.; McCormack, T.; Sasaki, T.; Okamura, Y. A voltage-sensing phosphatase, Ci-VSP, which shares sequence identity with PTEN, dephosphorylates phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. USA 2008, 105, 7970–7975.
- Huang, J.; Yan, J.; Zhu, S.; Wang, Y.; Shi, T.; Zhu, C.; Chen, C.; Liu, X.; Cheng, J.; Mustelin, T.; et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat. Commun. 2012, 3, 911.
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential Role for Nuclear PTEN in Maintaining Chromosomal Integrity. Cell 2007, 128, 157–170.
- Radu, A.; Neubauer, V.; Akagi, T.; Hanafusa, H.; Georgescu, M.-M. PTEN Induces Cell Cycle Arrest by Decreasing the Level and Nuclear Localization of Cyclin D1. Mol. Cell. Biol. 2003, 23, 6139–6149.
- Li, D.M.; Sun, H. PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15406–15411.
- Cheney, I.W.; Neuteboom, S.T.; Vaillancourt, M.T.; Ramachandra, M.; Bookstein, R. Adenovirus-mediated gene transfer of MMAC1/PTEN to glioblastoma cells inhibits S phase entry by the recruitment of p27Kip1 into cyclin E/CDK2 complexes. Cancer Res. 1999, 59, 2318–2323.
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220.
- Di Cristofano, A.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 1998, 19, 348–355.
- Alimonti, A.; Carracedo, A.; Clohessy, J.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458.
- Ortega-Molina, A.; Efeyan, A.; Lopez-Guadamillas, E.; Muñoz-Martin, M.; Gómez-López, G.; Cañamero, M.; Mulero, F.; Pastor, J.; Martinez, S.; Romanos, E.; et al. Pten Positively Regulates Brown Adipose Function, Energy Expenditure, and Longevity. Cell Metab. 2012, 15, 382–394.
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696.
- Zuo, Q.; Liu, J.; Huang, L.; Qin, Y.; Hawley, T.; Seo, C.; Merlino, G.; Yu, Y. AXL/AKT axis mediated-resistance to BRAF in-hibitor depends on PTEN status in melanoma. Oncogene 2018, 37, 3275–3289.
- Qin, Y.; Zuo, Q.; Huang, L.; Huang, L.; Merlino, G.; Yu, Y. PERK mediates resistance to BRAF inhibition in melanoma with impaired PTEN. NPJ Precis. Oncol. 2021, 5, 68.
- Whang, Y.E.; Wu, X.; Suzuki, H.; Reiter, R.E.; Tran, C.; Vessella, R.L.; Said, J.W.; Isaacs, W.B.; Sawyers, C.L. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc. Natl. Acad. Sci. USA 1998, 95, 5246–5250.
- McMenamin, M.E.; Soung, P.; Perera, S.; Kaplan, I.; Loda, M.; Sellers, W.R. Loss of PTEN expression in paraffin- 711 em-bedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999, 59, 4291–4296.
- Suzuki, H.; Freije, D.; Nusskern, D.R.; Okami, K.; Cairns, P.; Sidransky, D.; Isaacs, W.B.; Bova, G.S. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998, 58, 204–209.
- Choi, S.W.; Lee, Y.; Shin, K.; Koo, H.; Kim, D.; Sa, J.K.; Cho, H.J.; Shin, H.-M.; Lee, S.J.; Kim, H.; et al. Mutation-specific non-canonical pathway of PTEN as a distinct therapeutic target for glioblastoma. Cell Death Dis. 2021, 12, 374.
- Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068.
- Agahozo, M.C.; Van Bockstal, M.R.; Groenendijk, F.H.; Bosch, T.V.D.; Westenend, P.; Van Deurzen, C.H.M. Ductal carcinoma in situ of the breast: Immune cell composition according to subtype. Mod. Pathol. 2019, 33, 196–205.
- Khoury, K.; Tan, A.R.; Elliott, A.; Xiu, J.; Gatalica, Z.; Heeke, A.L.; Isaacs, C.; Pohlmann, P.R.; Schwartzberg, L.S.; Simon, M.; et al. Prevalence of Phosphatidylinositol-3-Kinase (PI3K) Pathway Alterations and Co-alteration of Other Molecular Markers in Breast Cancer. Front. Oncol. 2020, 10, 1475.
- Lazaridis, G.; Kotoula, V.; Vrettou, E.; Kostopoulos, I.; Manousou, K.; Papadopoulou, K.; Giannoulatou, E.; Bobos, M.; Sotiropoulou, M.; Pentheroudakis, G.; et al. Opposite Prognostic Impact of Single PTEN-loss and PIK3CA Mutations in Early High-risk Breast Cancer. Cancer Genom.-Proteom. 2019, 16, 195–206.
- Leskela, S.; Pérez-Mies, B.; Rosa-Rosa, J.M.; Cristobal, E.; Biscuola, M.; Palacios-Berraquero, M.L.; Ong, S.; Guia, X.M.-G.; Palacios, J. Molecular Basis of Tumor Heterogeneity in Endometrial Carcinosarcoma. Cancers 2019, 11, 964.
- McConechy, M.K.; Hoang, L.N.; Chui, M.H.; Senz, J.; Yang, W.; Rozenberg, N.; Mackenzie, R.; McAlpine, J.N.; Huntsman, D.G.; Clarke, B.A.; et al. In-depth molecular profiling of the biphasic components of uterine carcinosarcomas. J. Pathol. Clin. Res. 2015, 1, 173–185.
- Kaur, H.; Samarska, I.; Lu, J.; Faisal, F.; Maughan, B.L.; Murali, S.; Asrani, K.; Alshalalfa, M.; Antonarakis, E.S.; Epstein, J.I.; et al. Neuroendocrine differentiation in usual-type prostatic adenocarcinoma: Molecular characterization and clinical significance. Prostate 2020, 80, 1012–1023.
- Al Bashir, S.; Alzoubi, A.; Alfaqih, M.A.; Kheirallah, K.; Smairat, A.; Haddad, H.; Al-Dwairy, A.; Fawwaz, B.; Alzoubi, M.; Trpkov, K. PTEN Loss in a Prostate Cancer Cohort from Jordan. Appl. Immunohistochem. Mol. Morphol. AIMM 2020, 28, 389–394.
- Luongo, F.; Colonna, F.; Calapà, F.; Vitale, S.; Fiori, M.E.; De Maria, R. PTEN Tumor-Suppressor: The Dam of Stemness in Cancer. Cancers 2019, 11, 1076.
- Hollis, R.L.; Thomson, J.P.; Stanley, B.; Churchman, M.; Meynert, A.M.; Rye, T.; Bartos, C.; Iida, Y.; Croy, I.; Mackean, M.; et al. Molecular stratification of endometrioid ovarian carcinoma predicts clinical outcome. Nat. Commun. 2020, 11, 4995.
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576.
- Giles, K.M.; Rosenbaum, B.E.; Berger, M.; Izsak, A.; Li, Y.; Bochaca, I.I.; de Miera, E.V.-S.; Wang, J.; Darvishian, F.; Zhong, J.; et al. Revisiting the Clinical and Biologic Relevance of Partial PTEN Loss in Melanoma. J. Investig. Dermatol. 2019, 139, 430–438.
- Abe, I.; Lam, A.K.-Y. Anaplastic Thyroid Carcinoma: Current Issues in Genomics and Therapeutics. Curr. Oncol. Rep. 2021, 23, 31.
- Helderman, N.C.; Bajwa-Ten Broeke, S.W.; Morreau, H.; Suerink, M.; Terlouw, D.; van der Werf, A.S.; van Wezel, T.; Nielsen, M. The diverse molecular profiles of lynch syndrome-associated colorectal cancers are (highly) dependent on un-derlying germline mismatch repair mutations. Crit. Rev. Oncol./Hematol. 2021, 163, 103338.
- Xu, B.; Fuchs, T.L.; Dogan, S.; Landa, I.; Katabi, N.; Fagin, J.A.; Tuttle, R.M.; Sherman, E.J.; Gill, A.J.; Ghossein, R. Dissecting Anaplastic Thyroid Carcinoma: A Comprehensive Clinical, Histologic, Immunophenotypic, and Molecular Study of 360 Cases. Thyroid 2020, 30, 1505–1517.
- Podsypanina, K.; Ellenson, L.H.; Nemes, A.; Gu, J.; Tamura, M.; Yamada, K.M.; Cordon-Cardo, C.; Catoretti, G.; Fisher, P.E.; Parsons, R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc. Natl. Acad. Sci. USA 1999, 96, 1563–1568.
- Kwabi-Addo, B.; Giri, D.; Schmidt, K.; Podsypanina, K.; Parsons, R.; Greenberg, N.; Ittmann, M. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc. Natl. Acad. Sci. USA 2001, 98, 11563–11568.
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; de Boer, V.C.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic Elevation of PTEN Induces a Tumor-Suppressive Metabolic State. Cell 2012, 149, 49–62.
- Tsao, H.; Yang, G.; Goel, V.; Wu, H.; Haluska, F.G. Genetic Interaction Between NRAS and BRAF Mutations and PTEN/MMAC1 Inactivation in Melanoma. J. Investig. Dermatol. 2004, 122, 337–341.
- Ko, J.M.; Fisher, D.E. A new era: Melanoma genetics and therapeutics. J. Pathol. 2010, 223, 242–251.
- Chen, S.T.; Geller, A.C.; Tsao, H. Update on the Epidemiology of Melanoma. Curr. Dermatol. Rep. 2013, 2, 24–34.
- Stahl, J.M.; Cheung, M.; Sharma, A.; Trivedi, N.R.; Shanmugam, S.; Robertson, G.P. Loss of PTEN promotes tumor develop-ment in malignant melanoma. Cancer Res. 2003, 63, 2881–2890.
- Bonneau, D.; Longy, M. Mutations of the human PTEN gene. Hum. Mutat. 2000, 16, 109–122.
- Marsh, D.J.; Coulon, V.; Lunetta, K.; Rocca-Serra, P.; Dahia, P.L.M.; Zheng, Z.; Liaw, D.; Caron, S.; Duboué, B.; Lin, A.Y.; et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum. Mol. Genet. 1998, 7, 507–515.
- Torres, J.; Pulido, R. The Tumor Suppressor PTEN Is Phosphorylated by the Protein Kinase CK2 at Its C Terminus: Implications for pten stability to proteasome-mediated degradation. J. Biol. Chem. 2001, 276, 993–998.
- Lu, Y.; Yu, Q.; Liu, J.H.; Zhang, J.; Wang, H.; Koul, D.; McMurray, J.S.; Fang, X.; Yung, W.A.; Siminovitch, K.A.; et al. Src Family Protein-tyrosine Kinases Alter the Function of PTEN to Regulate Phosphatidylinositol 3-Kinase/AKT Cascades. J. Biol. Chem. 2003, 278, 40057–40066.
- Fragoso, R.; Barata, J.T. Kinases, tails and more: Regulation of PTEN function by phosphorylation. Methods 2015, 77, 75–81.
- Li, Z.; Dong, X.; Wang, Z.; Liu, W.; Deng, N.; Ding, Y.; Tang, L.; Hla, T.; Zeng, R.; Li, L.; et al. Regulation of PTEN by Rho small GTPases. Nat. Cell Biol. 2005, 7, 399–404.
- Yim, E.-K.; Peng, G.; Dai, H.; Hu, R.; Li, K.; Lu, Y.; Mills, G.B.; Meric-Bernstam, F.; Hennessy, B.T.; Craven, R.J.; et al. Rak Functions as a Tumor Suppressor by Regulating PTEN Protein Stability and Function. Cancer Cell 2009, 15, 304–314.
- Mulholland, D.J.; Dedhar, S.; Wu, H.; Nelson, C.C. PTEN and GSK3β: Key regulators of progression to androgen-independent prostate cancer. Oncogene 2006, 25, 329–337.
- Chen, J.-H.; Zhang, P.; Chen, W.-D.; Li, D.-D.; Wu, X.-Q.; Deng, R.; Jiao, L.; Li, X.; Ji, J.; Feng, G.-K.; et al. ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy 2015, 11, 239–252.
- Raftopoulou, M.; Etienne-Manneville, S.; Self, A.; Nicholls, S.; Hall, A. Regulation of Cell Migration by the C2 Domain of the Tumor Suppressor PTEN. Science 2004, 303, 1179–1181.
- Vazquez, F.; Ramaswamy, S.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN Tail Regulates Protein Stability and Function. Mol. Cell. Biol. 2000, 20, 5010–5018.
- Das, S.; Dixon, J.E.; Cho, W. Membrane-binding and activation mechanism of PTEN. Proc. Natl. Acad. Sci. USA 2003, 100, 7491–7496.
- Zhang, J.; Lee, Y.-R.; Dang, F.; Gan, W.; Menon, A.V.; Katon, J.M.; Hsu, C.-H.; Asara, J.M.; Tibarewal, P.; Leslie, N.R.; et al. PTEN Methylation by NSD2 Controls Cellular Sensitivity to DNA Damage. Cancer Discov. 2019, 9, 1306–1323.
- Kim, H.C.; Steffen, A.M.; Oldham, M.L.; Chen, J.; Huibregtse, J.M. Structure and function of a HECT domain ubiquitin-binding site. EMBO Rep. 2011, 12, 334–341.
- Wang, X.; Trotman, L.C.; Koppie, T.; Alimonti, A.; Chen, Z.; Gao, Z.; Wang, J.; Erdjument-Bromage, H.; Tempst, P.; Cordon-Cardo, C.; et al. NEDD4-1 Is a Proto-Oncogenic Ubiquitin Ligase for PTEN. Cell 2007, 128, 129–139.
- Lee, Y.R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.Y.; Fu, T.M.; Chen, H.; Ishikawa, T.; Chiang, S.Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364, eaau0159.
- Myers, M.P.; Pass, I.; Batty, I.H.; Van der Kaay, J.; Stolarov, J.P.; Hemmings, B.A.; Wigler, M.; Downes, C.P.; Tonks, N.K. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc. Natl. Acad. Sci. USA 1998, 95, 13513–13518.
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.-I.; Parsons, R.; Yamada, K.M. Inhibition of Cell Migration, Spreading, and Focal Adhesions by Tumor Suppressor PTEN. Science 1998, 280, 1614–1617.
- Dey, N.; Crosswell, H.E.; De, P.; Parsons, R.; Peng, Q.; Su, J.D.; Durden, D.L. The Protein Phosphatase Activity of PTEN Regulates Src Family Kinases and Controls Glioma Migration. Cancer Res. 2008, 68, 1862–1871.
- Bassi, C.; Ho, J.; Srikumar, T.; Dowling, R.J.O.; Gorrini, C.; Miller, S.J.; Mak, T.W.; Neel, B.G.; Raught, B.; Stambolic, V. Nuclear PTEN Controls DNA Repair and Sensitivity to Genotoxic Stress. Science 2013, 341, 395–399.
- Iijima, M.; Devreotes, P. Tumor Suppressor PTEN Mediates Sensing of Chemoattractant Gradients. Cell 2002, 109, 599–610.
- Funamoto, S.; Meili, R.; Lee, S.; Parry, L.; Firtel, R.A. Spatial and Temporal Regulation of 3-Phosphoinositides by PI 3-Kinase and PTEN Mediates Chemotaxis. Cell 2002, 109, 611–623.
- Gildea, J.J.; Herlevsen, M.; Harding, M.A.; Gulding, K.M.; Moskaluk, C.A.; Frierson, H.F.; Theodorescu, D. PTEN can inhibit in vitro organotypic and in vivo orthotopic invasion of human bladder cancer cells even in the absence of its lipid phosphatase activity. Oncogene 2004, 23, 6788–6797.
- Tamura, M.; Gu, J.; Takino, T.; Yamada, K. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: Differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 1999, 59, 442–449.
- Davidson, L.; Maccario, H.; Perera, N.M.; Yang, X.; Spinelli, L.; Tibarewal, P.; Glancy, B.; Gray, A.; Weijer, C.J.; Downes, C.P.; et al. Suppression of cellular proliferation and invasion by the concerted lipid and protein phosphatase activities of PTEN. Oncogene 2010, 29, 687–697.
- Innocenti, M.; Zucconi, A.; Disanza, A.; Frittoli, E.; Areces, L.B.; Steffen, A.; Stradal, T.E.B.; Di Fiore, P.P.; Carlier, M.-F.; Scita, G. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nature 2004, 6, 319–327.
- Leng, Y.; Zhang, J.; Badour, K.; Arpaia, E.; Freeman, S.; Cheung, P.; Siu, M.; Siminovitch, K. Abelson-interactor-1 promotes WAVE2 membrane translocation and Abelson-mediated tyrosine phosphorylation required for WAVE2 activation. Proc. Natl. Acad. Sci. USA 2005, 102, 1098–1103.
- Qi, Y.; Liu, J.; Chao, J.; Greer, P.A.; Li, S. PTEN dephosphorylates Abi1 to promote epithelial morphogenesis. J. Cell Biol. 2020, 219, e201910041.
- Qi, Y.; Liu, J.; Chao, J.; Scheuerman, M.P.; Rahimi, S.A.; Lee, L.Y.; Li, S. PTEN suppresses epithelial–mesenchymal transition and cancer stem cell activity by downregulating Abi1. Sci. Rep. 2020, 10, 12685.
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—an excellent servant but a bad master. J. Transl. Med. 2012, 10, 183.
- Bamburg, J.R.; McGough, A.; Ono, S. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol. 1999, 9, 364–370.
- Wioland, H.; Guichard, B.; Senju, Y.; Myram, S.; Lappalainen, P.; Jégou, A.; Romet-Lemonne, G. ADF/Cofilin Accelerates Actin Dynamics by Severing Filaments and Promoting Their Depolymerization at Both Ends. Curr. Biol. 2017, 27, 1956–1967.e7.
- Bamburg, J.R.; Minamide, L.S.; Wiggan, O.; Tahtamouni, L.H.; Kuhn, T.B. Cofilin and Actin Dynamics: Multiple Modes of Regulation and Their Impacts in Neuronal Development and Degeneration. Cells 2021, 10, 2726.
- Gu, T.; Zhang, Z.; Wang, J.; Guo, J.; Shen, W.H.; Yin, Y. CREB Is a Novel Nuclear Target of PTEN Phosphatase. Cancer Res. 2011, 71, 2821–2825.
- Shirao, T.; Sekino, Y. General Introduction to Drebrin. Adv. Exp. Med. Biol. 2017, 1006, 3–22.
- Grintsevich, E.E. Effects of neuronal drebrin on actin dynamics. Biochem. Soc. Trans. 2021, 49, 685–692.
- Dart, A.E.; Gordon-Weeks, P.R. The Role of Drebrin in Cancer Cell Invasion. Adv. Exp. Med. Biol. 2017, 1006, 375–389.
- Shnitsar, I.; Bashkurov, M.; Masson, G.R.; Ogunjimi, A.A.; Mosessian, S.; Cabeza, E.A.; Hirsch, C.L.; Trcka, D.; Gish, G.; Jiao, J.; et al. PTEN regulates cilia through Dishevelled. Nat. Commun. 2015, 6, 8388.
- Gu, J.; Tamura, M.; Pankov, R.; Danen, E.; Takino, T.; Matsumoto, K.; Yamada, K. Shc and Fak Differentially Regulate Cell Motility and Directionality Modulated by Pten. J. Cell Biol. 1999, 146, 389–404.
- Liliental, J.; Moon, S.Y.; Lesche, R.; Mamillapalli, R.; Li, D.; Zheng, Y.; Sun, H.; Wu, H. Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr. Biol. 2000, 10, 401–404.
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174.
- Tonsing-Carter, E.; Hernandez, K.M.; Kim, C.; Harkless, R.V.; Oh, A.; Bowie, K.; West-Szymanski, D.C.; Betancourt-Ponce, M.A.; Green, B.D.; Lastra, R.R.; et al. Glucocorticoid receptor modulation decreases ER-positive breast cancer cell proliferation and suppresses wild-type and mutant ER chromatin association. Breast Cancer Res. 2019, 21, 82.
- Obradovic, M.; Hamelin, B.; Manevski, N.; Couto, J.P.; Sethi, A.; Coissieux, M.-M.; Muenst, S.; Okamoto, R.; Kohler, H.; Schmidt, A.; et al. Glucocorticoids promote breast cancer metastasis. Nature 2019, 567, 540–544.
- Shi, Y.; Wang, J.; Chandarlapaty, S.; Cross, J.; Thompson, C.B.; Rosen, N.; Jiang, X. PTEN is a protein tyrosine phosphatase for IRS1. Nat. Struct. Mol. Biol. 2014, 21, 522–527.
- Bochman, M.L.; Schwacha, A. The Mcm2-7 Complex Has In Vitro Helicase Activity. Mol. Cell 2008, 31, 287–293.
- Labib, K.; Tercero, J.A.; Diffley, J.F.X. Uninterrupted MCM2-7 Function Required for DNA Replication Fork Progression. Science 2000, 288, 1643–1647.
- Blow, J.J.; Dutta, A. Preventing re-replication of chromosomal DNA. Nat. Rev. Mol. Cell Biol. 2005, 6, 476–486.
- Tenca, P.; Brotherton, D.; Montagnoli, A.; Rainoldi, S.; Albanese, C.; Santocanale, C. Cdc7 Is an Active Kinase in Human Cancer Cells Undergoing Replication Stress. J. Biol. Chem. 2007, 282, 208–215.
- Wozniak, D.J.; Kajdacsy-Balla, A.; Macias, V.; Ball-Kell, S.; Zenner, M.L.; Bie, W.; Tyner, A.L. PTEN is a protein phosphatase that targets active PTK6 and inhibits PTK6 oncogenic signaling in prostate cancer. Nat. Commun. 2017, 8, 1508.
- Abbas, A.; Romigh, T.; Eng, C. PTEN interacts with RNA polymerase II to dephosphorylate polymerase II C-terminal domain. Oncotarget 2019, 10, 4951–4959.
- Shinde, S.R.; Maddika, S. PTEN modulates EGFR late endocytic trafficking and degradation by dephosphorylating Rab7. Nat. Commun. 2016, 7, 10689.
- Brenner, W.; Schneider, E.; Keppler, R.; Prawitt, D.; Steinwender, C.; Roos, F.C.; Thüroff, J.W.; Lausch, E. Migration of renal tumor cells depends on dephosphorylation of Shc by PTEN. Int. J. Oncol. 2011, 38, 823–831.
- Patel, A.; Sabbineni, H.; Clarke, A.; Somanath, P.R. Novel roles of Src in cancer cell epithelial-to-mesenchymal transition, vascular permeability, microinvasion and metastasis. Life Sci. 2016, 157, 52–61.
- Zhang, S.; Huang, W.C.; Li, P.; Guo, H.; Poh, S.-B.; Brady, S.W.; Xiong, Y.; Tseng, L.-M.; Li, S.H.; Ding, Z.; et al. trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469.