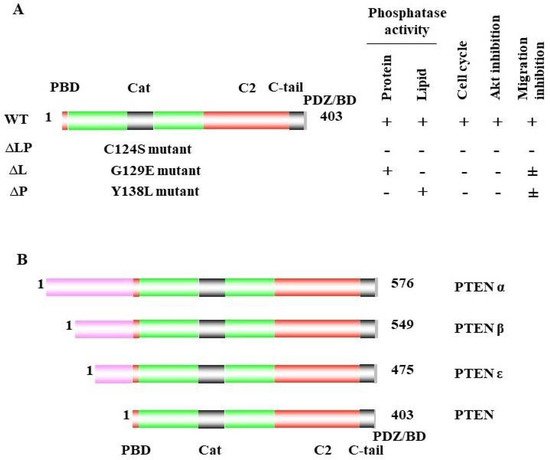
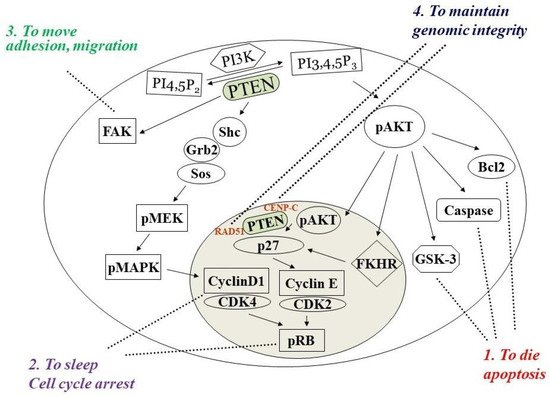
Phosphatase and tensin homolog deleted on chromosome ten (PTEN) is a multifunctional tumor suppressor with protein- and lipid-phosphatase activities. The inactivation of PTEN is commonly found in all human cancers and is correlated with tumor progression. PTEN-lipid-phosphatase activity has been well documented to dephosphorylate phosphatidylinositol-3, 4, 5-phosphate (PIP3), which hinders cell growth and survival by dampening the PI3K and AKT signaling activity. PTEN-protein-phosphatase activity dephosphorylates the different proteins and acts in various cell functions.
- PTEN
- PTEN lipid phosphatase
- PTEN protein phosphatase
- mutation
- PTEN protein substrate
- tumorigenesis
1. PTEN Dual Lipid and Protein Phosphatase
2. PTEN Inactivation in Cancer Progression
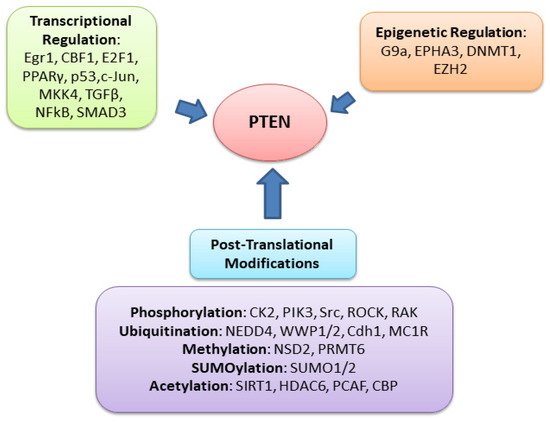
PTEN is one of the most highly mutated genes in human cancers. The dysfunction of PTEN by genetic mutation or epigenetic silencing contributes to most cancer development and progression (Figure 4, Table 1) [7][8][23][7,8,33]. It is often associated with high-grade and metastatic potential drug resistance [24][25][34,35] and poor patient prognosis [26][27][28][36,37,38]. The inactivation of PTEN is caused by various mechanisms, including genetic loss, point mutation, epigenetic regulation, and posttranslational modifications. Most alterations result in the loss of or reduction in PTEN protein.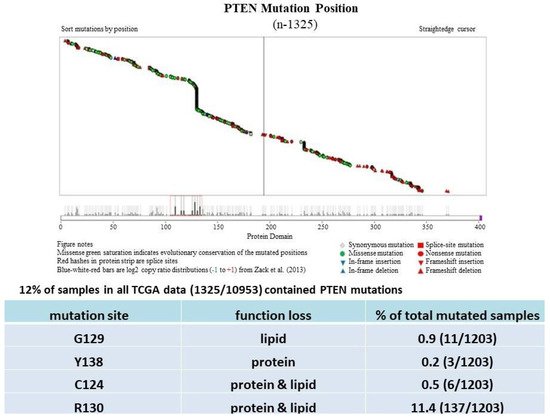
| Site/Tissue | Tumor Type | Range | Average | Comment | Reference(s) | |||
|---|---|---|---|---|---|---|---|---|
| Brain | Glioblastoma | 12–84% | 29% (88/303) | Mostly LOH | [29][30] | [53,54] | ||
| Breast | Ductal carcinoma | 11–55% | 33% (415/1257) | Mostly LOH | [31][32][33] | [55,56,57] | ||
| Endometrium | Endometrioid carcinoma | 19–82% | 67% (352/529) | LOH and mutation | [34] | [58 | [35] | ,59] |
| Prostate | Adenocarcinoma | 12–63% | 33% (88/267) | Mostly LOH | [36][37] | [60,61] | ||
| Ovary | Cystadenocarcinoma | 9–61% | 30% (33/112) | LOH and mutation | [38][39] | [62,63] | ||
| Skin | Melanoma | 11–39% | 30% (57/190) | Mostly LOH | [40][41] | [64,65] | ||
| Thyroid | Carcinoma (ATC) | 10–41% | 11% (21/196) | Mostly LOH | [42][43][44] | [66,67,68] |
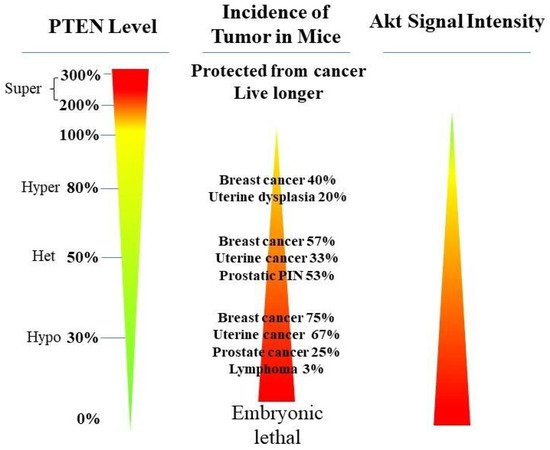
PTEN genetic loss is involved in various tumor types, such as glioblastoma, breast ductal carcinoma, endometrial carcinoma, prostate adenocarcinoma, ovary cystadenocarcinoma, melanoma, pancreatic adenocarcinoma, colorectal cancer, etc. (Table 1). In GEM mice, PTEN deficiency leads to embryonic lethality, and even a small reduction in the PTEN levels enhances the cancer incidence [20][21][45][46][23,24,39,40]. Contrarily, the systemic overexpression of PTEN in GEM models amplifies its tumor-suppressive function and protects against tumorigenesis [47][41], which suggests that the precise level of PTEN expression is a critical factor for the tumor-suppressor function, and that the reduction in PTEN activity is a driving mechanism for tumor progression. The pathologic characteristics that are associated with PTEN deletion in mice are phenocopied in a wide range of human tumors with loss-of-heterozygosity (LOH) mutations. In cancer, the PI3K/PTEN/Akt pathway has been identified as one of the critical molecular axes driving tumorigenesis [48][49][50][51][42,43,44,45]. The loss of PTEN activity has been reported to be responsible for many of the phenotypes of cancers, and it affects the development of 15–70% of human cancers [25][49][50][35,43,44] (Table 1).
Much has been learned about the features of PTEN point mutation through the analysis of PTEN germline mutations found in patients with Cowden disease (CD) [52][46]. Patients with CD, who harbor missense mutations in the PTEN phosphatase domain, are cancer-prone and develop more lesions than patients with PTEN deletion, which causes the complete loss of the PTEN function [53][47].
34. PTEN-Protein-Phosphatase Substrates and PI3K-Independent Function
3.1. PTEN
4.1. PTEN
PTEN has been reported to play a vital role in regulating cell migration, invasion, and metastasis [69][72][73][74][75][48,115,116,117,118]. As indicated above, the naturally derived PTEN point mutant G129E [9][75][76][9,118,119], which loses lipid but maintains protein-phosphatase activity, retains the ability to inhibit cell migration and invasion, as well as metastasis [69][48]. To study the other mechanisms involved in cell migration, Reftopoulou and colleagues found that PTEN protein phosphatase is required for migration. They further demonstrated that PTEN protein phosphatase dephosphorylates its C domain at Thr383, inhibiting the migration independent of its effects on the PI3K pathway [61][21].3.2. Abi1
4.2. Abi1
Abl-interactor 1 (Abi1) is a core component of the WASP-family verprolin homologous protein (WAVE) regulatory complex (WRC). Abi1 acts as a core scaffold protein to mediate the membrane recruitment and stabilization of WRC subunits [77][78][122,123], and it is regulated by extracellular cues and intracellular signaling pathways. PTEN can bind and dephosphorylate Abi1at Y213 and S216, triggering its degradation through the calpain pathway, thereby promoting epithelial differentiation and polarization [79][99], as well as epithelial–mesenchymal transition and cancer-stem-cell activity [80][124]. PTEN dephosphorylates Abi1 and downregulates the WRC at the cell cortex, thereby reorganizing the actin cytoskeleton to facilitate the formation of the apical actin belt and adherent junctions.3.3. Β-Catenin
4.3. Β-Catenin
Transforming growth factor β (TGF-β) is a pleiotropic cytokine that plays a role in growth suppression in normal epithelial cells, but it supports metastasis formation in many tumors [81][125].3.4. Cofilin-1
4.4. Cofilin-1
Cofilin-1 is an essential actin regulator. As an actin-depolymerizing factor (ADF)/cofilins family protein, Cofilin-1 regulates the rapid depolymerization of actin microfilaments that give actin its characteristic dynamic instability and its central role in muscle contraction, cell motility, and transcription regulation [82][83][84][126,127,128]. The activity of cofilin is regulated by a variety of mechanisms, including specific phosphorylation and dephosphorylation [84][128].3.5. CREB
4.5. CREB
The transcription factor cyclic AMP response element-binding protein (CREB) is activated via phosphorylation at serine133, which mediates gene transcription and promotes cell proliferation and survival. PTEN protein phosphatase is required for the dephosphorylation of CREB in the nucleus. Under PTEN-deficient conditions, CREB phosphorylation is enhanced independently of the PI3K/AKT pathway. The inhibition of the PI3K/AKT pathway does not affect the CREB phosphorylation in PTEN-deficient cells. C124S-mutant PTEN cannot dephosphorylate CREB, while G129E and wild-type PTEN can, which demonstrates that the protein-phosphatase activity of PTEN is essential for dephosphorylating and colocalizing with CREB in the nucleus [85][102], which suggests that PTEN regulates gene expression through this mechanism.3.6. Drebrin
4.6. Drebrin
Drebrin is a protein that is encoded by the DBN1 gene. It is a crucial regulator of the actin cytoskeleton in neuronal cells for synaptic plasticity, neurogenesis, and neuronal migration, as well as in cancer cells for tumor invasion [86][87][88][132,133,134]. The defect of Drebrin in expression and activation contributes to the pathogenesis.3.7. Dvl
4.7. Dvl
PTEN is an essential regulator of multicilia formation and cilia disassembly via Dishevelled (DVL2) phosphorylation. DVL is an important component of the WNT signaling pathways that plays a role during convergent extension movements. DVL is a ciliogenesis regulator in Xenopus and human epithelial cells, and it has been identified as a direct substrate for PTEN. Among the DVL proteins, DVL2 and DVL3 have the strongest associations with PTEN. The knockdown of PTEN increases the DVL2 phosphorylation on serine143 during cilia formation. By studying wild-type PTEN and mutants of PTEN (C124S, G129E, Y138L), it was confirmed that the protein-phosphatase activity of PTEN is responsible for directly dephosphorylating DVL2 on serine 143 [89][104], which implicates PTEN in multicilia formation and movement.3.8. FAK
4.8. FAK
Focal adhesion kinase (FAK) is one of the earliest confirmed substrates for PTEN. PTEN overexpression inhibits cell migration via reducing the phosphorylation of FAK. The effect of PTEN on cell spreading was examined on fibronectin (FN) in multiple cell lines: NIH 3T3 cells, human fibroblast cells, DBTRG-05MG cells (glioblastoma), and U-87MG (glioma) cells. The overexpression of PTEN delayed or inhibited the spreading in these cell lines [69][48]. FAK was first found to be a substrate of the Src proto-oncogene. It is an important regulator of cell adhesion and motility. The tyrosine phosphorylation of FAK is associated with focal contacts that form at ECM integrin junctions, and integrin-binding proteins recruit FAK to the focal contacts. It has been reported that PTEN suppresses cell migration, invasion, and metastasis through the dephosphorylation of FAK at Tyr397 [69][90][48,105]; however, specific FAK dephosphorylation sites have not been observed in other cell types [91][136], and other mechanistic details have yet to be uncovered.3.9. Glucocorticoid Receptor (GR)
4.9. Glucocorticoid Receptor (GR)
GR is a pleiotropic nuclear receptor and transcriptional regulatory factor that controls the network of glucocorticoid (GC)-responsive genes in a positive or negative manner for regulating numerous physiological and cellular processes [92][137]. In cancer, GR activation also appeared as tumor-suppressing [93][138] and tumor-promoting effects [94][139].