Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Vivi Li and Version 3 by Vivi Li.
Spine metastases are a common life-threatening complication of advanced-stage malignancies and often result in poor prognosis. Symptomatic spine metastases develop in the course of about 10% of malignant neoplasms. Therefore, it is essential for contemporary medicine to understand metastatic processes in order to find appropriate, targeted therapeutic options. Thanks to continuous research, there appears more and more detailed knowledge about cancer and metastasis, but these transformations are extremely complicated, e.g., due to the complexity of reactions, the variety of places where they occur, or the participation of both tumor cells and host cells in these transitions.
- spine
- metastasis
- tumor
- cancer
1. Introduction
Spine metastases are a common life-threatening complication of advanced-stage malignancies and often result in poor prognosis. Symptomatic spine metastases develop in the course of about 10% of malignant neoplasms [1].
The spine is the most frequent localization among the bone metastatic lesions and the third most common site of metastases after lungs and liver [2]. The thoracic region is the site of metastatic spine tumors in 60–70% of the cases, the lumbosacral spine (20–25%) and the cervical spine (10–15%) are less common [3]. In the analysis of CT scans, it has been observed that metastatic lesions typically occur in the posterior part of the vertebral body at first and then penetrate the pedicles [4]. Bone metastases most frequently develop from primary solid tumors (such as breast (70%), prostate (85%), lung (40%), and kidney (40%)) [5] (Figure 1). Women suffering from breast cancer with overexpression of the estrogen receptor or the progesterone-one receptor or HER2 triple-positive women and men with castration-resistant prostate cancer are most vulnerable to bone metastases [6].
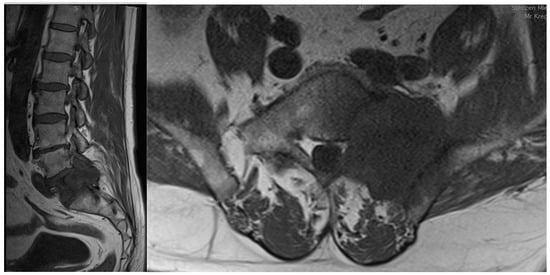
Figure 1. Sagittal and coronal T1-weighted MRI views of a patient diagnosed with sacral metastasis of a pulmonary squamous cell carcinoma.
Bone metastases can be osteolytic, osteoblastic, or mixed (osteolytic and osteoblastic components). According to Constans et al., osteolytic lesions constitute more than 70% of spinal metastases, 8% are osteoblastic, and mixed metastases (osteolytic and osteoblastic) account for 21% [7]. Osteolytic lesions are characteristic of metastases from breast cancer, lung cancer, and renal cancer, whereas metastases of prostate cancer most often form osteoblastic lesions. Causes of mixed lesions include many tumor types, but most frequently are observed in breast cancer [8].
Metastatic cells can reach the spine through different ways of dissemination—hematogenous (intravenous and arterial), by contiguity, and lymphatic. The intravenous route is the most common way of propagation and is carried out through the paravertebral venous plexus of Batson [9][10]. This venous system communicates the spine with intercostal veins of the pulmonary, caval, or portal systems and provides a direct route of dissemination for breast and prostate cancer to the spine [11].
The most common clinical symptom caused by metastatic lesions in spine is being refractory to severe treatment pain. Other symptoms include hypercalcemia, pathological fractures, and spinal cord or nerve root compression, which are referred to as skeletal-related events (SREs). The current treatment of spine metastases is mostly palliative. It focuses on the improvement of health-related quality of life (HRQoL) through the control of pain, protection, or improvement of neurological functions and maintenance of spinal stability [1]. To prevent SREs and increase the quality of life of patients with spinal metastases as a result, antiresorptive agents such as aminobisphosphonates and anti-RANKL monoclonal antibody denosumab have been approved by the FDA. These drugs act through inhibition of the osteoclast function, which leads to the interruption of the vicious cycle of bone metastases and increase of bone mass [12]. However, improvement of the overall survival (OS) and progression-free survival (PFS) by these agents suggested by some studies is highly questionable. Recently investigated novel therapeutic agents targeting specific molecular mechanisms in the bone microenvironment represent a promising way of treatment. The bone microenvironment constitutes a pivotal role in the process of spine metastases. Multiple molecular interactions between metastatic tumor cells and bone tissue result in the alteration of many molecular pathways, which leads to the development of osteolytic or osteoblastic lesions. Improving the understanding of these mechanisms may help in the development of novel drugs targeting specific biomarkers and pathways. Currently, multiple substances are being investigated in preclinical and clinical studies with regard to inhibition of the bone metastatic process.
2. Molecular Basis of Spine Metastases
2.1. Escape from the Primary Site
Tumor is a type of malignant cell growth where abnormal cells multiply uncontrollably with an ability for close or distant tissue invasion. Cells within a tumor may differ in many ways, e.g., in their proliferative potential or the ability to undergo apoptosis or metastasize [13][14][15][16]. The route that cancer cells must surmount to reach other organs is burdensome, with escape from the primary site being the first obstacle encountered [17][18][19]. Another essential step in the process of metastasis is the entrance of neoplastic cells into blood or lymphatic vessels through which they reach even remotely located organs [20]. Increased activity of the master regulator of angiogenesis—oxygen-sensitive transcriptional activator HIF (hypoxia-inducible factor-1)—contributes to both provision of the path utilized for metastatic spread as well as tumor cells’ nutrition via modulation of proangiogenic factors that include, e.g., VEGF (vascular endothelial growth factor), Ang-1, Ang-2 (angiopoietins), or PlGF (placental growth factor) [21][22][23]. A hallmark of metastasis, epithelial–mesenchymal transition (EMT) is a biological process where epithelial cells forfeit their adhesive properties and apical–basal polarity and acquire migratory as well as invasive features in order to transform into mesenchymal stem cells [24][25][26]. EMT is involved in both pathological (e.g., cancer formation) and physiological (embryogenesis, organ development, wound healing) processes [27][28] conditioned by biologically different EMT subtypes [29][30][31][32]. Reversibility of this process (MET—mesenchymal–epithelial transition) enables cancer cells to return to their original form in another organ [33][34]. Both EMT and MET comprise series of biochemical reactions regulated by a plethora of transcriptional factors including Snail/Slug, Twist, Six1, Cripto, TGF-β, and Wnt/β-catenin that, when activated, reprogram gene expression [35][36][37] and, in consequence, alter tumor cell properties. While the cytoskeleton undergoes remodeling, modified protein expression into proteins distinctive for mesenchymal cells, e.g., N-cadherin, FSP-1, α-SMA, α5β1 integrin, αvβ6 integrin, vimentin, type I collagen, laminin 5, and fibrotin lead to the termination of connection with the basal membrane [38][39][40][41]. As a result, cancer cells acquire enhanced capability for relocation. Additionally, enzymes produced by tumor cells that break down the extracellular matrix, MMP (matrix metalloproteinase) and ADAM (A disintegrin and metalloproteinase), enable vascular wall penetration [42][43]. EMT also provides resistance to apoptosis due to diminished cell adhesion. The same mechanism facilitates migration and, in turn, evasion of various factors that lead to cell destruction.2.2. Cancer Cells Dissemination
Cancers can spread to bones through various pathways, both venous and arterial, as well as through the lymphatic route or by direct contact [44]. Although the lymphatic system is mentioned as a potential route for spread, the main routes to enter the spinal column are comprised of venous and arterial vessels [45]. The Batson plexus, a network of veins devoid of valves that connect pelvic and thoracic with intraspinal veins contributes to spinal metastasis. Due to the absence of valves, any increase in vena cava pressure is followed by increased blood flow within the plexus, leading to cancer cells dissemination. In turn, neoplastic metastases reach the vertebral body directly through nutritional arteries [46][47]. Less frequently, neoplastic lesions metastasize through direct contact, e.g., prostate cancer that metastasizes to the lumbosacral spine [48]. Additionally, tumor cells have the ability to adhere to blood cells, including platelets, which serves as protection from the detrimental effects of hemodynamic forces during flow [49][50][51], as well as to bone marrow cells produced by tumor mimic precursors of immune cells, which aids to avoid innate immune response [52].2.3. Bone Invasion
There are two main theories of metastasis. One of them is the so called “seed and soil” theory proposed by Paget over 100 years ago. It says that cancers anchor where they find convenient conditions, similarly to seeds in fertile soil, a process not dependent on anatomical relations [52][53]. Ewing, in turn, said that metastases are only the result of the structure of the circulatory system and are strictly related to anatomical conditions, such as the diameter of the vessels or connections between organs. In the 1980s, the work by Hart and Fiedler on melanoma metastases confirmed that theory [54][55]. However, looking at the development of tumors, it seems that both theories are relevant. Bone marrow, due to abundant vascularization, constitutes a part of the bone tissue of relatively high affinity for tumor metastases. The bones of the axial skeleton (skull, spine, sternum, ribs, hips, shoulders) contain a substantial amount of red bone marrow and therefore are a frequent target of tumor spread [56]. Low velocity of the blood flow enables easier adhesion to endothelial cells and, in consequence, quicker integration with endosteum. Bone marrow and bone cells also produce cytokines, hormones, enzymes, as well as growth factors that regulate the immune system and affect the colonization of bone tissue by cancer cells [57][58]. Tumor cells release a plethora of factors [59][60], e.g., VEGFR1+—bone marrow-derived progenitor cells that activate VLA-4 that via binding to fibronectin [61][62][63][64] enables entrance to the potential metastatic site and create an appropriate environment, a premetastatic niche that facilitates tumor cells implantation [65][66][67][68][69]. The abovementioned mechanisms are best understood in an example of metastatic breast cancer. It has been found that CXCR4, also known as fusin, is necessary for breast cancer cells migration towards tissues that present a high quantity of its specific ligand—cytokine SDF1 (CXCL12). Among the organs that express high levels of SDF1 are lungs, liver, bone marrow, and brain, which explains the high affinity of breast cancer cells to these tissues [70][71][72][73]. Tyrosine kinase Src is activated through the binding of CXCL12 to CXCR4, and downstream effector AKT improves the survival of cancer cells that occupy bone tissues [74]. Primary tumor cells also produce substances that modify the extracellular matrix at sites of metastasis, e.g., lysyl oxidates. Conversely, exosomes and miRNAs are able to influence bone remodeling. Metalloproteinases (MMPs) and bone sialoproteins (BSPs) destroy basal membranes at the site of metastasis, stimulate angiogenesis, and activate various elements involved in the destruction of bone tissue and the spread of tumor cells [70][75][76][77]. Once cancer cells reach the bone marrow, their growth depends on multiple factors, including in situ vascularization, available space, type of bone remodeling, or proliferating potential of neoplastic cells [78]. Bone tissue is made up of three main types of cells: osteoblasts, osteoclasts, and osteocytes. Osteoclasts are cells that have the ability to dissolve and resorb bone tissue, while osteoblasts are responsible for the growth and remodeling of bone tissue. Processes that lead to the formation of bone metastases may have different mechanisms: osteolytic and osteoblastic. Sometimes both of these mechanisms work simultaneously. In a healthy organism, the activity of osteoclasts and osteoblasts corresponds to the RANK-RANKL/OPG system [79][80][81]. RANKL (receptor activator for nuclear factor κB ligand) is produced by the osteoblastic line and activated T lymphocytes [80][82]. It is responsible for activating the process of creating mature osteoclasts. While RANK (receptor activator for nuclear factor κB) is located on osteoclasts and serves as the main regulator during the formation of osteoclasts, RANK combines with RANKL, ligand of the receptor activator of nuclear factor kappa B (NF-κB), which at the same time causes upregulation of nuclear factor of activated T cells 1 (NFATc1) [83][84]. NFATc1 is the major regulator of cytokine expression in the process of osteoclastogenesis [85][86]. As a result of these changes, mature osteoblasts are formed. Their main task is old bone reabsorption, which causes the release of nutrients and creates space for osteoblasts. Osteoprotegrin (OPG) binds to RANK and blocks the formation of the RANK–RANKL complex, thereby inhibiting the maturation process of osteoclasts [87][88][89].2.4. Osteocyte Physiology and Pathology
Osteocytes have an impressive lifespan of up to 25 years, during which they undertake several important physiological functions. They differentiate from osteoblasts with four stages of formation, type I preosteocytes (osteoblastic osteocytes), type II preosteocytes (osteoid osteocytes), and type III preosteocytes (young and old osteocytes) [90]. During the process of bone formation, the osteoblastic cell body reduces in size, and its cytoplasm expands, springing processes from out of the cell’s wall. The Golgi apparatus during the type I and II cycles has to be well-developed to efficiently synthesize type I collagen essential for maintaining the bone matrix. Entering the type III preosteocyte phase, the Golgi apparatus is reduced in size, and the osteocyte matrix proceeds from the incompletely mineralized phase to the formation of old osteocytes with high mineral density [90]. Mature osteocytes express such markers as DMP1, Sost, as well as the cx43 protein, which is believed to be critical in the role of keeping the cell from entering apoptosis [90][91]. It is theorized that cx43 influences bone cell activity by regulating the osteoprotegerin and sclerostin levels [91].2.5. Osteoblast Physiology and Pathology
The aforementioned osteoblasts are generated from pluripotent mesenchymal stem cells that take on the crucial role of bone matrix synthesis by firstly establishing the collagen, OCN, osteonectin, BSP II, and osteopontin proteins alongside decorin and biglycan to create osteoids, which would be further mineralized [90]. Osteoblasts tend to communicate with osteocytes using the RANK–RANKL pathway as a way to order growth factor release from the bone matrix [92]. The aforementioned processes summarize the bone homeostasis. It has been established that the regions of the bone with the largest amount of turnover (trabecular bone) tend to become sites for metastatic cell growth. One very prominent factor of this cancer cell homing is CXC motif chemokine 12, also known as CXCL12 or SDF-1, produced by bone marrow stromal cells and osteoblasts, which was proven to be crucial to metastases [93][94]. Cancer cells interact with osteoblasts and osteoclasts, as well as the cytokines released by the bone in an otherwise physiological process [95].2.6. Osteoblastic Metastasis Pathogenesis
Bone tissue is considered to be the third place in the aspect of frequency of metastatic changes. Most of these metastatic changes are the result of oncological diseases, primarily breast and prostate cancer [8][96]. The statistics analyzed in previous years showed bone metastasis to be the effect of up to 70–75% of breast and prostate cancers [17]. A factor that one has to take into consideration is the bone’s extreme metabolic activity derived from the three main cell types: osteocytes, osteoblasts, and osteoclasts. Osteoblasts account for 4–6% of total cells, osteocytes—90–95%, osteoclasts—about 1–4% [90]. Osteoblastic metastasis is characterized by the deposition of new bone rather than lysis of the already existing structures. It is most notably present in prostate cancers, carcinoids, small-cell lung cancers, Hodgkin lymphomas, and medulloblastomas [17]. Tumor cells invading the bone tend to produce growth factors such as bone morphogenic proteins, epidermal growth factors, and platelet-derived growth factors. There have been consistent data proving that the physical contact of osteoblasts and prostate cancer cells promote tumor growth in vitro via protein ECM components, proteoglycans (PGs), and junction-related molecules [97]. Some of the more prominent factors of this process are BMPSs, TGF beta, and endothelin-1. BMP4 has been proven to stimulate osteoblast differentiation after being secreted from PCa-118b prostate cancer cells via the pSmad1–Notch–Hey1 and GSK3 β–β-catenin–Slug pathways [98]. The aforementioned TGF beta, specifically, TGF beta 2, is secreted from the prostate cancer metastasized cells [98] to foster the progression of tumor growth [99]. Another mechanism of growth is the secretion of endothelin-1 which downregulates DKK-1 and stimulates the secretion of the Wnt signaling pathways proven to be associated with lytic lesions and suppressing the growth of bone tissue in myelomas [100]. This occurs because DDK1 inhibits the production of osteoblasts by preventing the binding of low-density lipoprotein receptor-related proteins 5 and 6 (LRP5/6) in osteoblast precursors [101]. The role of PTHrP fragments in the process must not be underestimated. The parathyroid hormone-related protein increases calcium absorption and bone resorption [102], but it greatly increases the metastatic growth of cancer cells [103]. It has been theorized that NH2-terminal fragments of this protein stimulate bone formation via the ETA receptor because of the shared sequence homology to ET-1, which was proven to increase metastatic growth [104]. Other research has proven that PTHrP acts as a mediator in osteoblastogenesis, increasing early osteoblast differentiation and proliferation of bone marrow cells [105]. One other crucial aspect of bone metastasis that has to be taken into consideration is the so-called vicious cycle. Prostate cancer cells that have been freshly metastasized tend to produce PDGF, ET1, and BMPs that activate osteoblastic differentiation and bone matrix formation. As mentioned before, bone turnover marks the sites where tumors tend to grow, as the freshly synthesized structures are rich with growth factors such as IGF, FGF, and TGF-β that attract prostate cancer cells [95]. The physical contact of tumor cells and osteoblasts further promotes the secretion of growth factors—the vicious cycle continues to propel itself until it reaches the physical limits of the metastatic site.2.7. Osteolytic Bone Metastasis
When discussing osteolytic metastases, people must look at the research on breast cancer. The majority of breast cancer metastases are lytic; breast cancer cells produce TNF-A, IL-8, IL-11 [106], IGF1, LIF (leukemia inhibitory factor), lysyl oxidase [107], RANK ligands, and PTHrP (Figure 2) [108].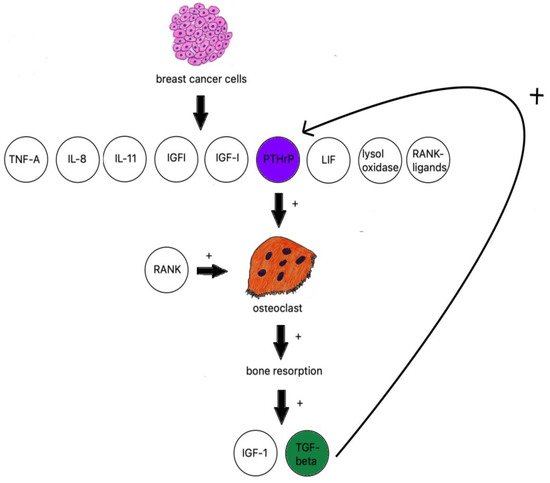
Figure 2. Breast cancer influence on osteoclastogenesis.
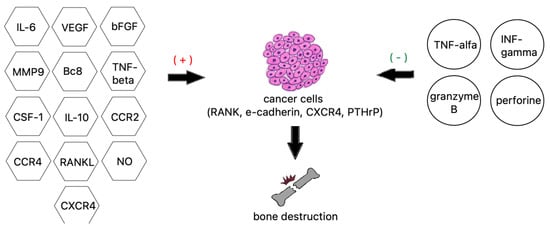
Figure 3. Factors contributing to bone destruction induced by metastatic cancer cells.
References
- Kurisunkal, V.; Gulia, A.; Gupta, S. Principles of Management of Spine Metastasis. Indian J. Orthop. 2020, 54, 181–193.
- Igoumenou, V.G.; Mavrogenis, A.F.; Angelini, A.; Baracco, R.; Benzakour, A.; Benzakour, T.; Bork, M.; Vazifehdan, F.; Nena, U.; Ruggieri, P.; et al. Complications of spine surgery for metastasis. Eur. J. Orthop. Surg. Traumatol. 2020, 30, 37–56.
- Tsukamoto, S.; Kido, A.; Tanaka, Y.; Facchini, G.; Peta, G.; Rossi, G.; Mavrogenis, A.F. Current Overview of Treatment for Metastatic Bone Disease. Curr. Oncol. 2021, 28, 3347–3372.
- Algra, P.R.; Heimans, J.J.; Valk, J.; Nauta, J.J.; Lachniet, M.; van Kooten, B. Do metastases in vertebrae begin in the body or the pedicles? Imaging study in 45 patients. AJR Am. J. Roentgenol. 1992, 158, 1275–1279.
- Ventura, C.; Núñez, S.V.; Gonçalves, A.; Abreu, C.; Costa, L. Bone Health in Metastatic Cancer. Semin. Oncol. Nurs. 2022, 38, 151278.
- Xiao, W.; Zheng, S.; Yang, A.; Zhang, X.; Zou, Y.; Tang, H.; Xie, X. Breast cancer subtypes and the risk of distant metastasis at initial diagnosis: A population-based study. Cancer Manag. Res. 2018, 10, 5329–5338.
- Constans, J.P.; de Divitiis, E.; Donzelli, R.; Spaziante, R.; Meder, J.F.; Haye, C. Spinal metastases with neurological manifestations. Review of 600 cases. J. Neurosurg. 1983, 59, 111–118.
- Coleman, R.E.; Croucher, P.I.; Padhani, A.R.; Clézardin, P.; Chow, E.; Fallon, M.; Guise, T.; Colangeli, S.; Capanna, R.; Costa, L.; et al. Bone metastases. Nat. Rev. Dis. Primers 2020, 6, 83.
- Ziu, E.; Viswanathan, V.K.; Mesfin, F.B. Spinal Metastasis. StatPearls. Published online March 3. Available online: https://www.ncbi.nlm.nih.gov/books/NBK441950/ (accessed on 6 August 2022).
- Maccauro, G.; Spinelli, M.S.; Mauro, S.; Perisano, C.; Graci, C.; Rosa, M.A. Physiopathology of Spine Metastasis. Int. J. Surg.Oncol. 2011, 2011, 107969.
- Papachristou, D.J.; Basdra, E.K.; Papavassiliou, A.G. Bone metastases: Molecular mechanisms and novel therapeutic interventions. Med. Res. Rev. 2012, 32, 611–636.
- Hofbauer, L.C.; Bozec, A.; Rauner, M.; Jakob, F.; Perner, S.; Pantel, K. Novel approaches to target the microenvironment of bone metastasis. Nat. Rev. Clin. Oncol. 2021, 18, 488–505.
- Homayoonfal, M.; Asemi, Z.; Yousefi, B. Potential anticancer properties and mechanisms of thymoquinone in osteosarcoma and bone metastasis. Cell. Mol. Biol. Lett. 2022, 27, 1–28.
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer Development, Progression, and Therapy: An Epigenetic Overview. Int. J. Mol. Sci. 2013, 14, 21087–21113.
- Biologii, P.; Tom, K. Przejście epitelialno-mezenchymalne w procesach nowotworzenia . Postępy Biologii Komórki 2018, 45, 223–236.
- Yin, W.; Wang, J.; Jiang, L.; James Kang, Y. Cancer and stem cells. Exp. Biol. Med. 2021, 246, 1791–1801.
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Gonçalves, F. Bone Metastases: An Overview. Oncol. Rev. 2017, 11, 321.
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28.
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027.
- Leong, S.P.; Naxerova, K.; Keller, L.; Pantel, K.; Witte, M. Molecular mechanisms of cancer metastasis via the lymphatic versus the blood vessels. Clin. Exp. Metastasis 2021, 39, 159–179.
- Font-Clos, F.; Zapperi, S.; la Porta, C.A.M. Blood Flow Contributions to Cancer Metastasis. iScience 2020, 23, 101073.
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10.
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed. Res. Int. 2015, 2015, 549412.
- Dudas, J.; Ladanyi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance. Cells 2020, 9, 428.
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773.
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412.
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506.
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587.
- Norgard, R.J.; Stanger, B.Z. Isolation and Identification of EMT Subtypes. Methods Mol. Biol. 2021, 2179, 315–326.
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4.
- Liao, T.T.; Yang, M.H. Revisiting epithelial-mesenchymal transition in cancer metastasis: The connection between epithelial plasticity and stemness. Mol. Oncol. 2017, 11, 792–804.
- Li, Q.; Hutchins, A.; Chen, Y.; Li, S.; Shan, Y.; Liao, B.; Zheng, D.; Shi, X.; Li, Y.; Chan, W.Y.; et al. A sequential EMT-MET mechanism drives the differentiation of human embryonic stem cells towards hepatocytes. Nat. Commun. 2017, 8, 15166.
- Ban, J.; Fock, V.; Aryee, D.N.T.; Kovar, H. Mechanisms, Diagnosis and Treatment of Bone Metastases. Cells 2021, 10, 2944.
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776.
- Basu, S.; Cheriyamundath, S.; Ben-Ze’ev, A. Cell–cell adhesion: Linking Wnt/β-catenin signaling with partial EMT and stemness traits in tumorigenesis. F1000Research 2018, 7, 1488.
- Zheng, M.; Jiang, Y.-P.; Chen, W.; Li, K.-D.; Liu, X.; Gao, S.-Y.; Feng, H.; Wang, S.-S.; Jiang, J.; Ma, X.-R.; et al. Snail and Slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget 2015, 6, 6794.
- Li, J.; Riedt, T.; Goossens, S.; García, C.C.; Szczepanski, S.; Brandes, M.; Pieters, T.; Dobrosch, L.; Gütgemann, I.; Farla, N.; et al. The EMT transcription factor Zeb2 controls adult murine hematopoietic differentiation by regulating cytokine signaling. Blood 2017, 129, 460–472.
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118.
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7, 1796.
- Huang, X.; Xiang, L.; Wang, B.; Hu, J.; Liu, C.; Ren, A.; Du, K.; Ye, G.; Liang, Y.; Tang, Y.; et al. CMTM6 promotes migration, invasion, and EMT by interacting with and stabilizing vimentin in hepatocellular carcinoma cells. J. Transl. Med. 2021, 19, 120.
- Huang, H.; Wright, S.; Zhang, J.; Brekken, R.A. Getting a grip on adhesion: Cadherin switching and collagen signaling. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 118472.
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370.
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83.
- Newland, R.; Chan, C.; Chapuis, P.; Keshava, A.; Rickard, M.; Stewart, P.; Suen, M.; Lee, K.; Dent, O. Relative effects of direct spread, lymph node metastasis and venous invasion in relation to blood borne distant metastasis present at the time of resection of colorectal cancer. Pathology 2020, 52, 649–656.
- Amelot, A.; Terrier, L.-M.; Cristini, J.; LeNail, L.-R.; Buffenoir, K.; Pascal-Moussellard, H.; Bonaccorsi, R.; Mathon, B. Approaching spinal metastases spread profile. Surg. Oncol. 2019, 31, 61–66.
- Kumar, N.; Tan, W.L.B.; Wei, W.; Vellayappan, B.A. An overview of the tumors affecting the spine—Inside to out. Neurooncol.Pract. 2020, 7 (Suppl. 1), i10.
- Chen, W.Z.; Shen, J.F.; Zhou, Y.; Chen, X.Y.; Liu, J.M.; Liu, Z.L. Clinical characteristics and risk factors for developing bone metastases in patients with breast cancer. Sci. Rep. 2017, 7, 11325.
- Klusa, D.; Lohaus, F.; Furesi, G.; Rauner, M.; Benešová, M.; Krause, M.; Kurth, I.; Peitzsch, C. Metastatic Spread in Prostate Cancer Patients Influencing Radiotherapy Response. Front. Oncol. 2020, 10, 627379.
- Schmied, L.; Höglund, P.; Meinke, S. Platelet-Mediated Protection of Cancer Cells from Immune Surveillance—Possible Implications for Cancer Immunotherapy. Front. Immunol. 2021, 12, 527.
- Liu, Y.; Zhang, Y.; Ding, Y.; Zhuang, R. Platelet-mediated tumor metastasis mechanism and the role of cell adhesion molecules. Crit. Rev. Oncol. Hematol. 2021, 167, 103502.
- Anvari, S.; Osei, E.; Maftoon, N. Interactions of platelets with circulating tumor cells contribute to cancer metastasis. Sci. Rep. 2021, 11, 15477.
- Akhtar, M.; Haider, A.; Rashid, S.; Al-Nabet, A.D.M.H. Paget’s “Seed and Soil” Theory of Cancer Metastasis: An Idea Whose Time has Come. Adv. Anat. Pathol. 2019, 26, 69–74.
- Liu, Q.; Zhang, H.; Jiang, X.; Qian, C.; Liu, Z.; Luo, D. Factors involved in cancer metastasis: A better understanding to “seed and soil” hypothesis. Mol. Cancer 2017, 16, 176.
- Pienta, K.J.; Robertson, B.A.; Coffey, D.S.; Taichman, R.S. The Cancer Diaspora: Metastasis beyond the seed and soil hypothesis. Clin. Cancer Res. 2013, 19, 5849–5855.
- Chernysheva, O.; Markina, I.; Demidov, L.; Kupryshina, N.; Chulkova, S.; Palladina, A.; Antipova, A.; Tupitsyn, N. Bone Marrow Involvement in Melanoma. Potentials for Detection of Disseminated Tumor Cells and Characterization of Their Subsets by Flow Cytometry. Cells 2019, 8, 627.
- Høilund-Carlsen, P.F.; Hess, S.; Werner, T.J.; Alavi, A. Cancer metastasizes to the bone marrow and not to the bone: Time for a paradigm shift! Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 893–897.
- Xiang, L.; Gilkes, D.M. The Contribution of the Immune System in Bone Metastasis Pathogenesis. Int. J. Mol. Sci. 2019, 20, 999.
- Li, H.; Wu, M.; Zhao, X. Role of chemokine systems in cancer and inflammatory diseases. MedComm 2022, 3, e147.
- Fathi, E.; Farahzadi, R.; Valipour, B.; Sanaat, Z. Cytokines secreted from bone marrow derived mesenchymal stem cells promote apoptosis and change cell cycle distribution of K562 cell line as clinical agent in cell transplantation. PLoS ONE 2019, 14, e0215678.
- Chen, P.; Wu, B.; Ji, L.; Zhan, Y. Cytokine Consistency Between Bone Marrow and Peripheral Blood in Patients with Philadelphia-Negative Myeloproliferative Neoplasms. Front. Med. 2021, 8, 598182.
- Florentin, J.; O’Neil, S.P.; Ohayon, L.L.; Uddin, A.; Vasamsetti, S.B.; Arunkumar, A.; Ghosh, S.; Boatz, J.C.; Sui, J.; Kliment, C.R.; et al. VEGF Receptor 1 Promotes Hypoxia-Induced Hematopoietic Progenitor Proliferation and Differentiation. Front. Immunol. 2022, 13, 882484.
- Ganta, V.C.; Choi, M.; Kutateladze, A.; Annex, B.H. VEGF 165 b Modulates Endothelial VEGFR1-STAT3 Signaling Pathway and Angiogenesis in Human and Experimental Peripheral Arterial Disease. Circ. Res. 2017, 120, 282–295.
- Massena, S.; Christoffersson, G.; Vågesjö, E.; Seignez, C.; Gustafsson, K.; Binet, F.; Hidalgo, C.H.; Giraud, A.; Lomei, J.; Weström, S.; et al. Identification and characterization of VEGF-A–responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood 2015, 126, 2016–2026.
- Marçola, M.; Rodrigues, C.E. Endothelial progenitor cells in tumor angiogenesis: Another brick in the wall. Stem Cells Int. 2015, 2015, 832649.
- Sanmartin, M.C.; Borzone, F.R.; Giorello, M.B.; Pacienza, N.; Yannarelli, G.; Chasseing, N.A. Bone marrow/bone pre-metastatic niche for breast cancer cells colonization: The role of mesenchymal stromal cells. Crit. Rev. Oncol. Hematol. 2021, 164, 103416.
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317.
- Jelgersma, C.; Vajkoczy, P. How to Target Spinal Metastasis in Experimental Research: An Overview of Currently Used Experimental Mouse Models and Future Prospects. Int. J. Mol. Sci. 2021, 22, 5420.
- Ren, G.; Esposito, M.; Kang, Y. Bone metastasis and the metastatic niche. J. Mol. Med. 2015, 93, 1203.
- Dong, Q.; Liu, X.; Cheng, K.; Sheng, J.; Kong, J.; Liu, T. Pre-metastatic Niche Formation in Different Organs Induced by Tumor Extracellular Vesicles. Front. Cell Dev. Biol. 2021, 9, 2643.
- Marques, C.S.; Santos, A.R.; Gameiro, A.; Correia, J.; Ferreira, F. CXCR4 and its ligand CXCL12 display opposite expression profiles in feline mammary metastatic disease, with the exception of HER2-overexpressing tumors. BMC Cancer 2018, 18, 741.
- Shi, Y.; Riese, D.J.; Shen, J. The Role of the CXCL12/CXCR4/CXCR7 Chemokine Axis in Cancer. Front. Pharmacol. 2020, 11, 1969.
- Zielińska, K.A.; Katanaev, V.L. The Signaling Duo CXCL12 and CXCR4: Chemokine Fuel for Breast Cancer Tumorigenesis. Cancers 2020, 12, 3071.
- Sun, Y.; Mao, X.; Fan, C.; Liu, C.; Guo, A.; Guan, S.; Jin, Q.; Li, B.; Yao, F.; Jin, F. CXCL12-CXCR4 axis promotes the natural selection of breast cancer cell metastasis. Tumour Biol. 2014, 35, 7765–7773.
- Xu, C.; Zhao, H.; Chen, H.; Yao, Q. CXCR4 in breast cancer: Oncogenic role and therapeutic targeting. Drug Des. Devel. Ther. 2015, 9, 4953–4964.
- Hamilton, S.L.; Ferando, B.; Eapen, A.S.; Yu, J.C.; Joy, A.R. Cancer Secretome May Influence BSP and DSP Expression in Human Salivary Gland Cells. J. Histochem. Cytochem. 2017, 65, 139–151.
- Zabkiewicz, C.; Resaul, J.; Hargest, R.; Jiang, W.G.; Ye, L. Bone morphogenetic proteins, breast cancer, and bone metastases: Striking the right balance. Endocr. Relat. Cancer 2017, 24, R349–R366.
- Rustamov, V.; Keller, F.; Klicks, J.; Hafner, M.; Rudolf, R. Bone sialoprotein shows enhanced expression in early, high- proliferation stages of three-dimensional spheroid cell cultures of breast cancer cell line MDA-MB-231. Front. Oncol. 2019, 9, 36.
- Chen, F.; Han, Y.; Kang, Y. Bone marrow niches in the regulation of bone metastasis. Br. J. Cancer 2021, 124, 1912–1920.
- Ming, J.; Cronin, S.J.F.; Penninger, J.M. Targeting the RANKL/RANK/OPG Axis for Cancer Therapy. Front. Oncol. 2020, 10, 1283.
- Sisay, M.; Mengistu, G.; Edessa, D. The RANK/RANKL/OPG system in tumorigenesis and metastasis of cancer stem cell: Potential targets for anticancer therapy. OncoTargets Ther. 2017, 10, 3801–3810.
- Litak, J.; Czyzewski, W.; Szymoniuk, M.; Pastuszak, B.; Litak, J.; Litak, G.; Grochowski, C.; Rahnama-Hezavah, M.; Kamieniak, P. Hydroxyapatite Use in Spine Surgery—Molecular and Clinical Aspect. Materials 2022, 15, 2906.
- de Groot, A.F.; Appelman-Dijkstra, N.M.; van der Burg, S.H.; Kroep, J.R. The anti-tumor effect of RANKL inhibition in malignant solid tumors—A systematic review. Cancer Treat. Rev. 2018, 62, 18–28.
- Sobacchi, C.; Menale, C.; Villa, A. The RANKL-RANK Axis: A Bone to Thymus Round Trip. Front. Immunol. 2019, 10, 629.
- Li, B.; Wang, P.; Jiao, J.; Wei, H.; Xu, W.; Zhou, P. Roles of the RANKL–RANK Axis in Immunity—Implications for Pathogenesis and Treatment of Bone Metastasis. Front. Immunol. 2022, 13, 922.
- Kim, K.J.; Lee, Y.; Hwang, H.G.; Sung, S.H.; Lee, M.; Son, Y.J. Betulin Suppresses Osteoclast Formation via Down-Regulating NFATc1. J. Clin. Med. 2018, 7, 154.
- Wang, L.; Chen, K.; He, J.; Kenny, J.; Yuan, Y.; Chen, J.; Liu, Q.; Tan, R.; Zhao, J.; Xu, J. Cytochalasin Z11 inhibits RANKL-induced osteoclastogenesis via suppressing NFATc1 activation. RSC Adv. 2019, 9, 38438–38446.
- Casimiro, S.; Vilhais, G.; Gomes, I.; Costa, L. The Roadmap of RANKL/RANK Pathway in Cancer. Cells 2021, 10, 1978.
- Wang, Y.; Liu, Y.; Huang, Z.; Chen, X.; Zhang, B. The roles of osteoprotegerin in cancer, far beyond a bone player. Cell Death Discov. 2022, 8, 252.
- Marley, K.; Bracha, S.; Seguin, B. Osteoprotegerin activates osteosarcoma cells that co-express RANK and RANKL. Exp. CellRes. 2015, 338, 32–38.
- Tresguerres, F.G.F.; Torres, J.; López-Quiles, J.; Hernández, G.; Vega, J.A.; Tresguerres, I.F. The osteocyte: A multifunctional cell within the bone. Ann. Anat. 2020, 227, 151422.
- Davis, H.M.; Pacheco-Costa, R.; Atkinson, E.G.; Brun, L.R.; Gortazar, A.R.; Harris, J.; Hiasa, M.; Bolarinwa, S.A.; Yoneda, T.; Ivan, M.; et al. Disruption of the Cx43/miR21 pathway leads to osteocyte apoptosis and increased osteoclastogenesis with aging. Aging Cell 2017, 16, 551–563.
- Ottewell, P.D. The role of osteoblasts in bone metastasis. J. Bone Oncol. 2016, 5, 124.
- Floranović, M.P.; Veličković, L.J. Effect of CXCL12 and Its Receptors on Unpredictable Renal Cell Carcinoma. Clin. Genitourin. Cancer 2020, 18, e337–e342.
- Hiraga, T. Bone metastasis: Interaction between cancer cells and bone microenvironment. J. Oral. Biosci. 2019, 61, 95–98.
- Kolb, A.D.; Shupp, A.B.; Mukhopadhyay, D.; Marini, F.C.; Bussard, K.M. Osteoblasts are “educated” by crosstalk with metastatic breast cancer cells in the bone tumor microenvironment. Breast Cancer Res. 2019, 21, 31.
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176.
- Suhovskih, A.V.; Kashuba, V.I.; Klein, G.; Grigorieva, E.V. Prostate cancer cells specifically reorganize epithelial cell-fibroblast communication through proteoglycan and junction pathways. Cell Adhes. Migr. 2017, 11, 39.
- Yu, G.; Shen, P.; Lee, Y.-C.; Pan, J.; Song, J.H.; Pan, T.; Lin, S.-C.; Liang, X.; Wang, G.; Panaretakis, T.; et al. Multiple pathways coordinating reprogramming of endothelial cells into osteoblasts by BMP4. iScience. 2021, 24, 102388.
- Ungefroren, H. Autocrine TGF-β in Cancer: Review of the Literature and Caveats in Experimental Analysis. Int. J. Mol. Sci. 2021, 22, 977.
- Wei, X.F.; Chen, Q.L.; Fu, Y.; Zhang, Q.K. Wnt and BMP signaling pathways co-operatively induce the differentiation of multiple myeloma mesenchymal stem cells into osteoblasts by upregulating EMX2. J. Cell. Biochem. 2019, 120, 6515–6527.
- Role of the Wnt/β-Catenin Signaling Pathway in Regulating the Phenotypic Transformation of Aortic Valvular Myofibroblasts to Osteoblast-like Cells. Available online: http://caod.oriprobe.com/articles/48652138/Role_of_the_Wnt_%CE%B2_catenin_signaling_pathway_in_regulating_the_phenotyp.htm (accessed on 6 August 2022).
- Winter, E.M.; Appelman-Dijkstra, N.M. Parathyroid Hormone-Related Protein-Induced Hypercalcemia of Pregnancy Successfully Reversed by a Dopamine Agonist. J. Clin. Endocrinol. Metab. 2017, 102, 4417–4420.
- Goltzman, D. Nonparathyroid Hypercalcemia. Front. Horm. Res. 2019, 51, 77–90.
- Vičić, I.; Belev, B. The pathogenesis of bone metastasis in solid tumors: A review. Croat. Med. J. 2021, 62, 270.
- Kamalakar, A.; Washam, C.L.; Akel, N.S.; Allen, B.J.; Williams, D.K.; Swain, F.L.; Leitzel, K.; Lipton, A.; Gaddy, D.; Suva, L.J. PTHrP(12-48) Modulates the Bone Marrow Microenvironment and Suppresses Human Osteoclast Differentiation and Lifespan. J. Bone Miner Res. 2017, 32, 1421–1431.
- Liang, M.; Ma, Q.; Ding, N.; Luo, F.; Bai, Y.; Kang, F.; Gong, X.; Dong, R.; Dai, J.; Dai, Q.; et al. IL-11 is essential in promoting osteolysis in breast cancer bone metastasis via RANKL-independent activation of osteoclastogenesis. Cell Death Dis. 2019, 10, 353.
- Reynaud, C.; Ferreras, L.; Di Mauro, P.; Kan, C.; Croset, M.; Bonnelye, E.; Pez, F.; Thomas, C.; Aimond, G.; Karnoub, A.E.; et al. Lysyl oxidase is a strong determinant of tumor cell colonization in bone. CancerRes. 2017, 77, 268–278.
- le Pape, F.; Vargas, G.; Clézardin, P. The role of osteoclasts in breast cancer bone metastasis. J. Bone Oncol. 2016, 5, 93–95.
- Brook, N.; Brook, E.; Dharmarajan, A.; Dass, C.R.; Chan, A. Breast cancer bone metastases: Pathogenesis and therapeutic targets. Int. J. Biochem. Cell Biol. 2018, 96, 63–78.
- Zheng, X.; Kang, W.; Liu, H.; Guo, S. Inhibition effects of total flavonoids from Scutellaria barbata D. Don on human breast carcinoma bone metastasis via down-regulating PTHrP pathway. Int. J. Mol. Med. 2018, 41, 3137–3146.
- Martin, T.J.; Johnson, R.W. Multiple actions of parathyroid hormone-related protein in breast cancer bone metastasis. Br. J.Pharmacol. 2021, 178, 1923–1935.
- Cheng, J.N.; Frye, J.B.; Whitman, S.A.; Kunihiro, A.G.; Pandey, R.; Funk, J.L. A Role for TGFβ Signaling in Preclinical Osteolytic Estrogen Receptor-Positive Breast Cancer Bone Metastases Progression. Int. J. Mol. Sci. 2021, 22, 4463.
- Wu, C.; Chen, M.; Sun, Z.; Ye, Y.; Han, X.; Qin, Y.; Liu, S. Wenshen Zhuanggu formula mitigates breast cancer bone metastasis through the signaling crosstalk among the Jagged1/Notch, TGF-β and IL-6 signaling pathways. J. Ethnopharmacol. 2019, 232, 145–154.
- Meng, X.; Ark, A.V.; Lee, P.; Hostetter, G.; Bhowmick, N.A.; Matrisian, L.M.; Williams, B.O.; Miranti, C.K.; Li, X. Myeloid-specific TGF-β signaling in bone promotes basic-FGF and breast cancer bone metastasis. Oncogene 2016, 35, 2370–2378.
- Das, D.; Hong, J. Prostaglandin E2 Receptor 4 (EP4): A Promising Therapeutic Target for the Treatment of Cancer and Inflammatory Diseases. Curr. Chem. Biol. 2020, 15, 50–68.
- Johnston, K.A.; Lopez, K.M. Lysyl oxidase in cancer inhibition and metastasis. Cancer Lett. 2018, 417, 174–181.
- Vaziri, N.; Shariati, L.; Javanmard, S.H. Leukemia inhibitory factor: A main controller of breast cancer. J. Biosci. 2020, 45, 143.
- Wang, M.; Wang, M.; Wang, Z.; Yu, X.; Song, Y.; Wang, C.; Xu, Y.; Wei, F.; Zhao, Y.; Xu, Y. Long non-coding RNA-CTD-2108O9.1 represses breast cancer metastasis by influencing leukemia inhibitory factor receptor. Cancer Sci. 2018, 109, 1764–1774.
- Hu, G.-F.; Wang, C.; Wu, G.; Zhang, C.; Zhu, W.; Chen, C.; Gu, Y.; Zhang, H.; Yang, Z. AZD3463, an IGF-1R inhibitor, suppresses breast cancer metastasis to bone via modulation of the PI3K-Akt pathway. Ann. Transl. Med. 2020, 8, 336.
- Okamoto, K.; Takayanagi, H. Osteoimmunology. Cold Spring Harb. Perspect. Med. 2019, 9, a031245.
More