Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Hong-Xi Xu and Version 3 by Jessie Wu.
Inflammatory bowel diseases (IBDs) are characterized by chronic inflammatory disorders that are a result of an abnormal immune response mediated by a cytokine storm and immune cell infiltration. Proinflammatory cytokine therapeutic agents, represented by tumor necrosis factor (TNF) inhibitors, have developed rapidly over recent years and are promising options for treating IBD. Antagonizing interleukins, interferons, and Janus kinases have demonstrated their respective advantages in clinical trials and are candidates for anti-TNF therapeutic failure. Furthermore, the blockade of lymphocyte homing contributes to the excessive immune response in colitis and ameliorates inflammation and tissue damage.
- cytokines
- IBD
- interleukins
1. Introduction
Cytokines are pivotal influencers of gut homeostasis and perpetual inflammation [1][6]. The appearance of anti-TNF drugs has drastically altered the therapeutic algorithm for IBD, yet a significant proportion of patients receiving anti-TNF therapy suffer from nonresponse or secondary loss of response. Fortunately, new treatment alternatives have been proactively explored (Figure 1), among which some have entered clinical practice and some have shown good efficacy in preclinical studies [2][7].
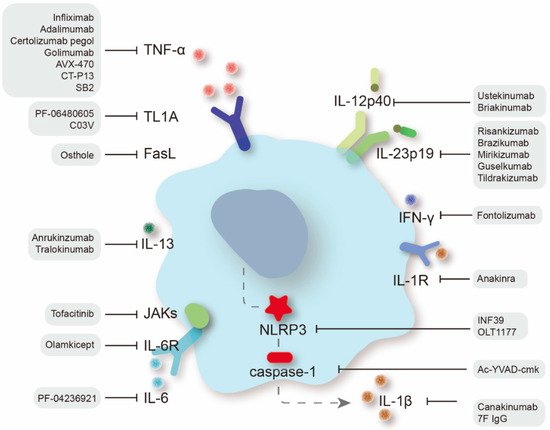
Figure 1.
Therapeutic targets of proinflammatory cytokines in IBD treatment and specific therapeutic agents.
2. Tumor Necrosis Factor Superfamily
The tumor necrosis factor superfamily (TNFSF) members are molecules with broad-spectrum activity and are well known to have pathological effects on IBD, the best known of which is TNF. The majority of TNFSF members are expressed by immune cells and modulate the homeostasis of T-cell-mediated immune responses [3][8]. Numerous recent studies have demonstrated that certain TNFSF members, including TNF-α, TL1A, FasL, and others, promote IBD pathogenesis through the enhancement of T-cell proinflammatory function and through the direct disruption of intestinal epithelial integrity [4][9].
2.1. Anti-TNF-α Agents
Infliximab (IFX) is an immunoglobulin G1 monoclonal antibody (mAb) with a high affinity for TNF-α that was initially licensed in 1998 by the FDA for treating patients with refractory active IBD, pioneering the era of biologics for IBD [5][6][10,11]. IFX is potent in both attaining and sustaining clinical remission and is also recommended for patients with IBD tha are unresponsive to 5-ASAs or corticosteroid therapy. A multiple-center randomized controlled trial (RCT) of pediatric CD patients [7][12] showed that first-line IFX intervention outperformed routine treatment in terms of short-term clinical (59% vs. 34%) and endoscopic remission (59% vs. 17%). Sequentially, more anti-TNF mAbs, such as adalimumab, certolizumab pegol, and golimumab, have been developed and licensed, showing promising clinical efficacy [8][13]. For example, in a multicenter, prospective cohort study of CD patients, approximately two-thirds of patients receiving adalimumab had a good prognosis and long-term maintenance of remission, reducing the incidence of colectomy [9][14]. In addition to intravenous monotherapy, a new oral polyclonal anti-TNF antibody, AVX-470, prepared from the colostrum of TNF-immune cows, showed efficacy comparable to that of oral prednisolone and parenteral etanercept in murine experimental colitis models and minimized systemic exposure to anti-TNF agents [10][15]. The first RCT of AVX-470 for UC demonstrated a good safety profile and was beneficial for refractory UC, with clinical, endoscopic, and biomarker (serum CRP and IL-6) improvements [11][16]. Additionally, TNF-related biosimilars, such as CT-P13 and SB2, have been developed to mitigate the surge in therapeutic costs caused by biological treatments [12][17], increasing the accessibility of appropriate biologic treatments by patients. Currently, over 20 biosimilars of infliximab and adalimumab are under development and have been described in detail [13][18].
2.2. Anti-TL1A Therapy
TNFSF member 15 gene (TNFSF15, also named TL1A) variants are correlated with the risk of IBD, which impacts the production of multiple cytokines to fuel mucosal inflammation [14][19]. TL1A is overexpressed in inflamed mucosa and is associated with CD fibrosis [15][20]. As TL1A is upstream of the proinflammatory process, anti-TL1A regimens may offer an attractive option for individuals unresponsive to TNF-α inhibitors and are a candidate for resolving currently nonreversible intestinal fibrosis. TL1A-overexpressing mice developed idiopathic ileitis with increased proximal colitis-inducing injury and fibrosis, whereas TL1A receptor-deficient mice exhibited resistance to a transmigration model of colitis and had reduced inflammation and intestinal fibrosis [16][21]. A human immunoglobulin G1 mAb against TL1A, PF-06480605, demonstrated improved endoscopic healing (38.2% at week 14, p < 0.001, and 95% CI = 23.82–53.68) and a favorable safety profile among individuals with refractory UC [17][22], probably in association with inflammatory T cells and fibrosis pathways [18][23]. C03V, a high affinity and selective human antibody to TL1A that neutralizes the biological activity of TL1A, remarkably attenuated the pathology of colitis triggered by trinitrobenzene sulfonic acid (TNBS) and alleviated fibrosis [19][24].
2.3. Targeting the FasL/FAS System
Mutations in TNFRSF6 (also named FasL) and its receptor FAS cause autoimmune lymphoid tissue proliferation syndrome and are involved in lymphocyte variation in IBD [20][25]. A marked increase in FasL-containing cells was observed in active UC patients in comparison to either remission or control groups based on immunohistochemistry of the colonic mucosa [21][26]. Conversely, a study on an acute colitis rat TNBS model discovered that isolated dendritic cells from mesenteric lymph nodes of wild-type rats expressed more FasL than those from colitis rats, and exogenous infusion of DCs genetically engineered to overexpress FasL decreased T-cell IFN-γ production and enhanced T-cell apoptosis, effectively reducing colonic inflammation [22][27]. Osthole restored the downregulation of Fas and FasL expression in bone marrow mesenchymal stem cells (BMSCs) caused by immune impairment and restored the ability of BMSCs to induce T-cell apoptosis and immunosuppressive function over experimental colitis [23][28].
2.4. Other TNFSF Members with Potential Efficacy
We are gaining insight into the value of other members of TNFSF in the IBD process as research progresses. TNFSF14, also named LIGHT, resides on T cells and participates in their activation, and its transgenic expression on murine T cells drives pathogenic inflammation in several organs, including the intestine [24][29]. Mice with Tnfsf14 deficiency were found to undergo more serious colitis and have lower survival rates than wild-type mice, suggesting that LIGHT may mediate innate immune activities and the resilience of gut inflammation by transmitting signals through lymphotoxin β receptors in the colon [25][30]. Furthermore, single-cell analysis of colonic mesenchymal cells in UC patients also revealed a subpopulation of activated mesenchymal populations with TNFSF14 expression, which is assumed to limit colonic epithelial proliferation and impede wound repair responses [26][31]. TNFSF10, also named TRAIL, mediates autoimmune inflammation and cellular homeostasis by transmitting apoptotic signals to induce apoptosis and was found to suppress autoimmune colitis by inhibiting colonic T-cell activation [27][32].
It is worth noting that not every patient responds to anti-TNF therapy and many suffer from the underlying opportunistic infections that come with treatment. High-dose administration regimens are recommended by experts to address the insufficient efficacy of anti-TNFs, and individualized dosing strategies are encouraged to be implemented with drug monitoring in clinical practice to avoid inadequate blockade of TNF in serum and tissues [28][33]. Natural products and herbs have also shown modulation of TNF-α activity in experimental models and clinical trials, which might be an effective intervention to bypass the side effects of specific anti-TNF therapy [29][34]. Natural polyphenols from plants, such as curcumin [30][35], mangiferin [31][36], and catechin [32][37], have demonstrated anti-inflammatory effects in TNBS/DSS-induced colitis models and reduced TNF-α expression. In addition, complementing anti-TNF therapy with nutritional biocompounds, such as antioxidant-enriched purple corn supplement [33][38], has exhibited beneficial effects on the induction and maintenance of IBD remission. While studies on natural products are currently mostly preclinical, their potential for the treatment and prevention of IBD deserves constant attention.
3. Interleukins
Multiple interleukins secreted by lymphocytes mediate their pro/anti-inflammatory effects, and inhibition of proinflammatory cytokines (IL-6, IL-1β, IL-12, and IL-23) is an effective strategy to ameliorate colitis. Presently, agents targeting interleukins have exhibited some advantages over anti-TNF agents.
3.1. Anti-IL-12/IL-23 Agents
IL-12 and IL-23, produced mainly by inflammatory myeloid cells, are highly expressed in colitis patients and induce differentiation and responses in TH1 cells and IL-17-producing T helper cells (TH17), respectively [34][39]. IL-12 and IL-23, as heterodimeric cytokines, are formed by the homogeneous subunit p40 in conjunction with p35 and p19, respectively. The roles of IL-12 and IL-23 are temporally distinct in the progression of IBD, where IL-12 is engaged in the early stages of colitis by activating TH1 cells and macrophages, while IL-23 shapes the chronic exacerbation of the disease [35][40].
Antagonizing p40, the cosubunit for IL-12 and IL-23, is a potent blockade of disease development. Ustekinumab (UST), a mAb against the p40 subunit, has already been licensed for CD treatment. In two 8-week RCTs of intravenous induction therapy for intermediate-to-severe active CD patients (UNITI-1 and UNITI-2), patients undergoing either primary or secondary nonresponse to TNF antagonists achieved significantly higher remission rates in patients receiving intravenous UST than those receiving placebo (in UNITI-1, 34.3% vs. 21.5%; in UNITI-2, 51.7% vs. 28.7%). A double-blind, active comparative phase 3b RCT (SEAVUE) involving multiple countries showed that 124 (65%) moderate-to-severe CD patients (n = 191) without biologic therapy remained in clinical remission after UST treatment at week 52 [36][41]. Moreover, patients taking maintenance doses of UST injections every 8 or 12 weeks achieved prolonged disease remission, and the occurrence of undesirable events was comparable to the placebo [37][42]. UST is equally effective in inducing and sustaining remission for patients with moderate-to-severe UC compared with placebo [38][43], and future studies are expected to compare the effectiveness of UST with existing biologics in treating UC [39][44]. Briakinumab, an anti-IL-12p40 mAb, has been terminated from production and follow-up studies due to uncertainty and low confidence in its actual role in the induction and maintenance of clinical remission in CD patients [40][45].
IL-23 is a pivotal promoter for chronic intestinal inflammation, and targeting IL-23-specific p19 subunits has proven to be effective in phase 2 studies of IBD [41][46], which may facilitate selective blockade of intestinal lymphatic trafficking without compromising systemic immune defense [42][47]. There are four mAbs (risankizumab, brazikumab, mirikizumab, and guselkumab) currently applied in clinical trials for either CD or UC [43][48]. Risankizumab showed curative efficacy and an admissible safety profile in patients suffering moderate-to-severe CD [44][49], and subcutaneous risankizumab demonstrated maintenance efficacy (including >71% clinical relief and >42% endoscopic remission) in a phase 2 study with good treatment tolerability [45][50]. Additionally, IL-23/IL-17 axis-related genes were differentially expressed in the transcriptome profile of the ileum and colon of patients after risankizumab treatment [46][51]. Brazikumab treatment was correlated with clinical improvement in patients with moderate-to-severe CD at 8 and 24 weeks [47][52]. Mirikizumab exhibited an effective induction of a positive clinical response following 12 weeks of treatment in UC patients (22.6% in the 200 mg group vs. 4.8% in the placebo group) and showed lasting benefits during the entire maintenance period, while it should be noted that the treatment effect of mirikizumab did not seem to proceed in a dose-dependent manner, and further studies are required to determine the optimal dose [48][53]. Comparatively, approximately 50% of patients initially unresponsive to mirikizumab achieved a clinical response after receiving a prolonged induction period at an increased dose (600 or 1000 mg [49][54]). Different doses of guselkumab had a good safety profile compared to placebo, achieving better clinical and endoscopic remission (200 mg: 57.4%, 500 mg: 55.6%, and 1200 mg: 45.9% vs. placebo: 16.4%) in a phase 2 clinical trial (GLAXI-1). Additionally, tildrakizumab, a high affinity humanized IgG1κ antibody targeting IL23p19, has proven efficacy in chronic plaque psoriasis and is expected to be implemented into IBD management [50][55].
Since the effect of IL-23 blockers in the management of chronic inflammatory diseases was first confirmed in psoriasis and subsequently revalidated in other inflammatory diseases, such as IBD, learning from the long-term experience of their applications in other diseases may help to accelerate the optimization of IL-12/23 dosing regimens for IBD patients by gastroenterologists and avoid drug adverse events [51][56].
3.2. IL-6/IL-6R Inhibitors
The engagement of IL-6 in IBD and colorectal cancer pathogenesis has been well explored [52][57], and some anti-IL-6 mAbs have been tested in RCTs against autoimmune diseases. IL-6 is rapidly produced in response to tissue injury and infection and promotes host immunity by stimulating acute phase responses and immune reactivity, while imbalanced IL-6 synthesis contributes to the pathological effects on chronic inflammation and immune action [53][58]. IL-6 mediates the proliferation of TH17 cells in concert with TGF-β by affecting signal transducers and activators of transcription (STAT3), which is essential for TH17 cell maintenance and function [54][59].
PF-04236921, an antibody against IL-6, was investigated among adult CD patients (ANDANTE I and II), with an increased remission rate after a 12-week treatment period (27.4% in PF-04236921 vs. 10.9% in placebo) [55][60]. PF-04236921 has the potential to induce clinical responses and palliations for refractory CD patients not responding to anti-TNF regimens, but its possible side effects (gastrointestinal abscesses and abdominal pain) warrant attention during subsequent development. Clone MP5-20F3 (an IL-6 mAb) neutralized DSS-induced elevated IL-6 concentrations and inhibited Claudin-2 expression in mice, improving intestinal inflammation and permeability, and its therapeutic effects were further amplified upon coadministration with an anti-TNF-α mAb [56][61]. An intriguing study on diet and colon cancer demonstrated that a high-calorie diet elevated IL-6 expression in porcine colonic mucosa and changed the expression of IL-6-associated proteins such as PI3KR4, IL-1α, and Map2k1, while a whole food diet inhibited HCD-induced alterations in IL-6-related proteins and modulation of IL-6 signaling was strongly associated with diet-related colitis/cancer [57][62]. Notably, IL-6 is involved in conditioning critical pharmacokinetic enzymes, such as cytochrome P450s (CYPs), and receiving anti-IL-6 management may restrict the AUCs of drugs with CYP substrates [58][63], which would impair drug management of many diseases.
Blocking IL-6R also effectively restricts IL-6 complex formation and modulates IL-6-mediated chronic inflammation. Tocilizumab, a humanized IL-6R blocker, is currently approved as a treatment for rheumatoid arthritis and has proven effective in other recalcitrant autoimmune diseases, and its therapeutic applications in IBD are promising [59][64]. Furthermore, since the chronic proinflammatory activity of IL-6 depends on the gp130 coreceptor for trans-signaling, the decoy protein sgp130Fc (olamkicept) was developed to specifically block IL-6 trans-signaling. Olamkicept resulted in a clinical response by 44% of patients with active IBD in a prospective 2a trial, and its clinical effectiveness was consistent with transcriptional changes in target inhibition [60][65].
Overall, IL-6 inhibition is a viable treatment option for IBD, but the benefit of completely blocking IL-6 or related receptors is restricted by its strong immunosuppression [60][65].
3.3. IL-1β/IL-1R Antagonists
IL-1β is the main proinflammatory mediator of inflammation. Its secretion is triggered predominantly by macrophages in response to injurious stimuli and it has high expression in IBD patients. The analysis of intestinal biopsy specimens and circulating cytokine profiles from 30 UC patients showed that nearly three-quarters of primary nonresponders to anti-TNF therapy were characterized by hyperexpression of IL-1β in serum and gut tissue [61][66]. Despite the fact that IL-1β is considered to be as crucial a cytokine as TNF-α in the onset of IBD, there are currently few specific antibodies against IL-1β, excluding canakinumab [62][67]. The absence of binding to murine-derived IL-1β hindered preclinical studies of canakinumab [63][68].
A novel chimeric anti-IL-1β-specific mAb, 7F IgG, with high affinity for various mammalian IL-1βs, blocked IL-1β to regulate proinflammatory cytokines and showed anticolitis activity in a TNBS-induced murine model [63][68]. Antagonism of the IL-1 receptor (IL-1R) is a therapeutic alternative to reduce the IL-1β stimulatory response. Endogenous IL-1R antagonists have been discovered to be important in host defense against overwhelming endotoxin-induced injury, and they are generated in experimental animal models of multiple diseases and in human autoimmune inflammatory diseases and are essential natural anti-inflammatory proteins in colitis [64][69]. Anakinra (an IL-1R antagonist) treatment ameliorated dinitrobenzene sulfonate (DNBS)-induced colitis in terms of macroscopic and histological changes, inflammatory cell infiltration, and oxidative stress [65][70]. Two IL10R-deficient patients with severe refractory IBD had significant clinical, endoscopic, and histological responses following 4–7 weeks of anakinra therapy [66][71]. A phase 2, multicenter RCT (IASO) of short-term anakinra treatment in patients with acute severe ulcerative colitis (ASUC) has been approved by the Cambridge Central Ethics Committee, a clinical trial has been authorized [67][72], and the prognostic outcome of using anakinra to intervene in IL-1 signaling in patients with ASUC is of ongoing interest. A proportion of de-N-acetylated oligomers (13 < dp < 20) were reported to rescue inflammatory impairments via an IL-1Ra-dependent pathway, and water-soluble galactosaminogalactan oligosaccharides might be therapeutically applicable as new anti-inflammatory glycosides in IL-1-mediated hyperinflammatory diseases [68][73].
Indeed, the release of IL-1β is driven by the activated NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, which is harmful in colitis [69][74]. In a comparative preclinical study of NLRP3 inflammasome signaling inhibitors, it was found that administration of INF39 (an NLRP3 inflammasome irreversible direct blocker), Ac-YVAD-cmk (a caspase-1 inhibitor), and anakinra to rats with DNBS-induced colitis resulted in colitis relief and that INF39 was more effective in reducing organizational increments of myeloperoxidase, TNF, and IL-1β, and attenuating intestinal inflammation [70][75]. OLT1177, a selective inhibitor of NLRP3 inflammasomes, was administered during the induction period of DSS-induced mice to alleviate the disease phenotype, and OLT1177 was effective in preventing the onset of DSS colitis. However, the administration of OLT1177 during the recovery phase of colitis did not promote the recovery course or tissue remission [70][75]. Curcumin potently inhibited NLRP3 inflammasome activation to alleviate colitis in a DSS-induced murine model, and the therapeutic outcome was partially limited following the use of MCC950 (a specific NLRP3 blocker) [71][76].
3.4. Other Interleukin Neutralizers with Potential Efficacy
Experimental animal studies and human genetic studies are gradually revealing the importance of the interleukin superfamily in regulating inflammation and tissue damage in the intestinal mucosa [72][77]. More members of the interleukin family are being noticed in colitis, and targeted therapies are being explored.
IL-18 is a proinflammatory interleukin matured by NLRP3 inflammasome activation, and IL-18 equilibrium in the epithelium has been implicated in barrier function in colitis, where IL-18 coordinates goblet cell maturation transcriptional programs [73][78]. Mokry conducted a Mendelian randomization study of 12,882 cases and 21,770 controls to examine the effect of elevated IL-18 on IBD susceptibility, and the results revealed that each genetically predicted SNP change (rs385076, rs17229943, and rs71478720) in IL-18 was associated with elevated susceptibility to IBD [74][79]. GSK1070806, a neo-IgG1 mAb that neutralizes IL-18, has been tested in a pilot trial in type 2 diabetes and kidney transplantation [75][80], and a corresponding RCT based on genomic detection of IL-18 in IBD is warranted.
Overproduction of IL-13 in active UC causes disturbances in the intestinal epithelium and may promote fibrosis. Anrukinzumab is a humanized IgG1 antibody conjugated to IL-13 that impedes the binding of IL-13 and IL-4Rα without affecting the attachment of IL-13 to IL-13α1/α2. After receiving anrukinzumab intravenously, the total serum level of IL-13 increased (free IL-13 binds to anrukinzumab) in patients with active UC, while changes in fecal calprotectin at 14 weeks posttreatment were not significantly different compared to placebo, and the treatment effect did not have a statistically significant difference [76][81]. Tralokinumab, an IL-13 neutralizing antibody, failed to induce a clinical response when used as an add-on therapy randomized to ambulatory adult UC patients, whereas tralokinumab had higher rates of clinical remission (18% vs. 6%) and mucosal healing (32% vs. 20%) than placebo, suggesting that it may benefit some UC patients and be well tolerated [77][82]. Likewise, although IL-17A is an active cytokine in gut inflammation, the clinical outcome of anti-IL-17A therapy (secukinumab and ixekizumab, anti-IL-17A mAbs) in CD patients is not favorable [78][83], and a study based on IL-17a-/- mice found that blockade of IL-17A may cause IL-6 upregulation and RORγt+ ILC recruitment during chronic colitis, leading to elevated IL-22 and even worsened disease [79][84].
Apart from the interleukins mentioned above, the role played by other interleukins, such as IL-5 [80][85], IL-21 [81][86], and IL-33 [82][87], in IBD development is being uncovered as the disease mechanism comes to be better understood, and subsequent drug development for them is anticipated. The network of interleukin family actions is intertwined, and a complete blockade of a singular interleukin may lead to systemic involvement. This may explain the involvement of some interleukin family members in mediating IBD and the lack of efficacy in targeted antagonistic therapy [83][88]. Long-term follow-up after anti-interleukin control and regular monitoring of relevant therapeutic indicators are necessary to avoid potential adverse drug effects.